Inborn errors of metabolism consist of a heterogeneous group of
disorders with multi-organ manifestations, including the heart. Although
they are individually rare and incidence data is difficult to collect,
they may be quite common collectively [1]. As the heart is a
metabolically active organ,it can be adversely affected by metabolic
defects [2]; the reported incidence of cardiac involvement varies from
15% to 60% [3-5]. To our knowledge there is no systemic review of the
cardiovascular manifestations of inborn errors of metabolism so far.
This narrative review aims to describe the cardiovascular
manifestations, during childhood, of common congenital metabolic
diseases.
A comprehensive literature review was conducted by
two independent reviewers using Pubmed (www.ncbi.nlm.nih.gov/pubmed)
as the medical database source and without applying any restrictions to
study design. Papers published in the last 20 years (including citations
of relevant articles found within) written in English, French, and
German were considered.
The terms used were ‘inborn errors of metabolism’,
‘metabolic defects’, ‘child’, ‘heart’, ‘cardiac’, ‘mitochondrial
disorders’, ‘carnitine’, ‘fatty acid metabolism’, ‘acidemia’, ‘storage
disorders’, ‘Pompe’, ‘Fabry’, ‘Barth syndrome’, ‘Smith-Lemli-Opitz’,
‘congenital disorders of glycosylation’, ‘cardiomyopathy’, ‘arrhythmia’,
‘heart rhythm disorders’, ‘valve’, ‘congenital heart disorders’, and
‘structural heart disorders’.
Our search initially identified 94 articles excluding
studies conducted solely in adult populations, describing only vascular
complications, or consisting of expert opinions or duplicate records.
Overall, 17 original papers (clinical or experimental studies), 5
reviews and 28 case reports or case series were identified.
Inborn Errors of Metabolism
Inborn errors of metabolism are traditionally
classified as urea cycle defects and disorders of carbohydrate meta-bolism,
amino acid metabolism, organic acid metabolism, fatty acid oxidation,
mitochondrial metabolism, peroxisomal function, porphyrine metabolism,
purine and pyrimidine metabolism, steroid metabolism, lysosomal storage,
and cholesterol biosynthesis [6].
In most cases, the underlying mechanism includes
mutations in genes coding for proteins, which are involved in metabolic
pathways. These changes may lead to abnormalities in synthesis or
catabolism of various substances, as well as to accumulation of
products, that are either toxic or interfere with normal body functions.
Various types of inheritance are present, although the majority of
inborn errors of metabolism are inherited in an autosomal recessive way
[7].
Pathophysiology of Cardiac Involvement
Cardiac manifestations among these patients include
cardiomyopathy (hypertrophic, dilated, restrictive), heart rhythm
disorders, valvular defects, and congenital heart structure disorders.
It should be noted; however, that more than one pattern of cardiac
involvement may be present in some inborn errors of metabolism (Table
I) [4,5,8-22].
TABLE I Major Cardiac Involvement in Common Metabolic Errors
| Type of
inborn error of metabolism |
Cardiomyopathy |
Heart rhythm
disorders |
Valvular
disease |
| Carnitine
deficiency [10-12,22 ] |
+++ |
++ |
+ |
| Fatty acid
oxidation disorders [8,9] |
+++ |
+++ |
- |
| Organic
acidemias17-21 ] |
+++ |
+ |
- |
| Storage
disorders [5, 13-16 ] |
+++ |
+++ |
+++ |
| Congenital
glycosylation disorders [4] |
+++ |
- |
- |
|
+++: Retrospective/prospective studies, ++: Many case
reports/series, +: Isolated case reports. |
In many cases, cardiac manifestations dominate the
clinical phenotype, as they include one of the prominent symptoms (e.g.,
Pompe disease or disorders of fatty acid oxidation). In other metabolic
defects; however, heart problems consist of minor symptoms and are
incidentally revealed during routine multisystem evaluation (e.g.,
glycogen storage disorders, mucopolysaccharidoses). Lastly, there are
cases of metabolic errors in which the heart may be the only affected
organ (e.g., some mitochondrial disorders) [2] (Table
II).
Table II Cardiac Manifestations in Inborn Errors of Metabolism
| Disease |
Prominent
finding |
Secondary
finding(s) |
Age at onset |
| Carnitine
deficiency |
Cardiomyopathy |
Heart
rhythm/ valvular
|
Neonatal to
early childhood |
|
|
disorders |
|
| Fatty acid
oxidation disorder |
Cardiomyopathy, heart |
– |
Neonatal to
early
|
|
rhythm
disorders |
|
childhood |
| Acidemias |
Cardiomyopathy |
Heart rhythm
disorders |
Neonatal to
childhood |
| Glycogen
storage disorders* |
Cardiomyopathy, valvular |
Heart rhythm
disorders |
Late infancy
to childhood
|
|
disorders |
|
|
| Pompe |
Cardiomyopathy, heart |
– |
Infancy to
childhood
|
|
rhythm/
valvular disorders |
|
|
| Gaucher* |
Cardiomyopathy, valvular |
Heart rhythm
disorders |
Late infancy
to childhood
|
|
disorders |
|
|
|
Mucopolysaccharidoses* |
Cardiomyopathy, valvular
|
Heart rhythm
disorders |
Late infancy
to childhood |
|
|
disorders |
|
| Congenital
glycosylation disorders |
Cardiomyopathy |
– |
Neonatal to
early childhood |
|
*Cardiac manifestations ate usually not a presenting feature. |
Pathophysiology includes three basic mechanisms: (i)
impaired energy production due to enzyme deficiency, disturbed transport
of molecules or cellular organelles dysfunction (e.g.,
mitochondrial dysfunction), (ii) infiltration of cardiac myocytes
with stored substrate and subsequent cellular damage, (iii)
accumulation of intermediary metabolites, which exert a toxic effect on
surrounding tissues and lead myocytes to apoptosis [2] (Fig. 1).
It is noteworthy that in many cases more than one mechanisms may be
involved, especially in later stages of the disease course.
Web
Table I summarizes the main cardiac manifestations of inborn
errors of metabolism found in the literature [3,5,13-19,22,23].
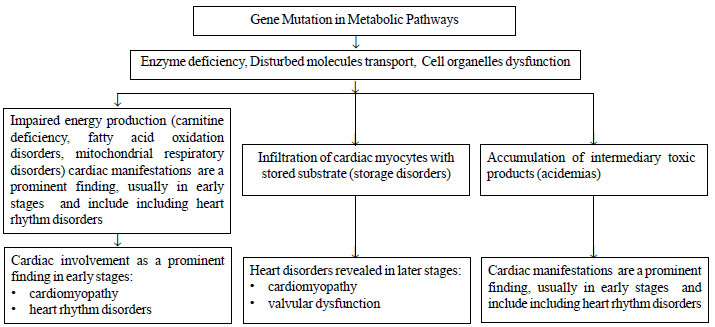 |
|
Fig. 1 Pathophysiological mechanisms
of cardiac involvement in metabolic disorders.
|
Impaired Energy Production
Disturbed energy production is the most prominent
underlying mechanism of cardiac involvement in carnitine
deficiency, fatty acid oxidation disorders, and other mitochondrial
disorders. Cardiac manifestations typically appear early in the course
of the disease, have an acute onset and dominate the clinical phenotype.
Furthermore, their identification often leads to the diagnosis of the
underlying defect [2].
It is estimated that inherited metabolic disorders
account for approximately 30% of definable causes of cardiomyopathy in
childhood [23]. More specifically, cardiomyopathy is the most common
clinical manifestation in children with primary carnitine deficiency and
includes dilated cardiomyopathy and hypertrophic cardio-myopathy. The
average age of cardiomyopathy appearance is 2-4 years of age, indicating
that it takes a long time for the changes in heart to manifest in severe
carnitine deficiency. While the incidence of dilated cardiomyopathy
seems to be higher than hypertrophic cardiomyopathy, a mild degree of
ventricular hypertrophy may be present in some patients presenting with
dilated cardiomyopathy [24,25].
Cardiomyopathy has also been reported in fatty acid
metabolism disorders. Defects involving oxidation of long or very long
chain fatty acids are more frequently associated with cardiomyopathy
than those involving oxidation of the short chain fatty acids. In fact,
most experimental and clinical studies have been conducted in this group
of patients. Very-long-chain acyl-CoA dehydrogenase (VLCAD) catalyzes
the first step in the beta-oxidation spiral of fatty acid metabolism
with infantile hypertrophic cardiomyopathy being the most common
clinical phenotype of its deficiency [8].
With regards to other energy production defects,
MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis and
stroke-like episodes) is associated with hypertrophic, dilated
cardiomyopathy and even with cases of left ventricular non-compaction.
However, very few cases of cardiomyopathy associated with MELAS in
pediatric or adolescent populations are found in literature [26].
Hypertrophy of left ventricle is the dominant pattern of myocardial
involvement in MERRF (myoclonic epilepsy with red-ragged fibers)
syndrome, in Leigh disease, as well as in complex I-V deficiency [2].
The incidence of these cases in childhood is extremely low.
Congenital disorders of glycosylation are also of
special interest. They represent a group of recently described
multisystem disorders characterized by defects in protein glycosylation.
Hypertrophic cardiomyopathy contributes significantly to the high
mortality of these patients, particularly those with subtype Ia (caused
by mutations in phosphomannomutase 2 gene). Cases of hypertrophic
cardiomyopathy and other cardiac related adverse events (cardiac
failure, tamponade, pericardial effusions) in this group have been
reported from the prenatal period and neonatal age to late childhood
[27,28]. On the other hand, dilated cardiomyopathy has been observed in
few subtypes of glycosylation disorders. It usually results in lethal
outcome and has been associated with mutations in dolichol kinase 1
gene. Therefore, patients with congenital disorders of glycosylation
type Ia should be monitored regularly by echocardiography for cardiac
complications. Furthermore, children with an undiagnosed cardiomyopathy
should be screened for glycosylation disorders [28].
Disturbed energy production is also involved in heart
rhythm disorders. Case reports describing prolonged QTc and ventricular
tachycardia in neonates and infants with fatty acid metabolism disorders
(very long and medium chain acyl-CoA dehydrogenase deficiency) have been
found. In many of these cases, heart rhythm disorder is the major
presenting symptom leading to the diagnosis of the underlying inborn
error [9,29,30] (Table II). Similarly, cases of QTc
prolongation and heart rhythm disorders in children with carnitine
deficiency have been identified in literature [10,11,31]. Studies have
also shown that specific mutations in mitochondrial DNA (e.g.,
G13513A mutation) are associated with increased risk for
Wolff-Parkinson-White syndrome in patients with MELAS syndrome or Leigh
disease [12].
With regards to other heart-related manifestations,
Trivellato, et al. [32] had described low plasma and urinary
carnitine levels in adult patients with idiopathic mitral valve
prolapse, but no further information on this topic is available. Mitral
valve regurgitation has been reported in a case of mitochondrial
cardiomyopathy [33]. Although dysfunction of mitochondria in patients
with valvular disorders has been histopathologically confirmed and
associated with aging, no correlation between specific mitochondrial
diseases and valvular defects has been reported [34].
Infiltration of Cardiac Myocytes With Stored
Substrate
Progressive infiltration of cardiac myocytes with
stored substrate is the basic pathophysiological mechanism in storage
disorders. Although hypertrophic cardio-myopathy is a well-recognized
manifestation in these disorders, there is poor literature documentation
related to cases specific to various subforms [2] (Table I).
In the vast majority of the published cases, cardiac hypertrophy is an
echocardiographic finding in asymptomatic patients and is related to the
natural course of the disease. Pompe disease and Anderson-Fabry disease,
in which cardiac hypertrophy usually occurs in the late childhood
period, present the only exceptions to the above [2] (Table II).
Most infants with Pompe disease develop
cardiomyopathy (massive hypertrophy of both ventricles) before the age
of 6 months and often present symptoms of congestive heart failure [35].
On the other hand, children with late-onset Pompe disease experience
slower progression of muscle involvement and do not usually have
significant cardiac manifestations [13]. In Fabry disease, left
ventricular hypertrophy is the most common pattern of cardiac
involvement in childhood and can appear at an early age in both genders
[2,14].
Symmetrical hypertrophy of the left ventricle is the
most frequent echocardiographic finding in glycogen storage disease type
III [15]. However, according to Mogahed, et al. [15], there is no
relation between skeletal myopathy and cardiomyopathy.
Arrhythmias can also appear in later stages of
storage disorders secondary to progressive heart dysfunction, and do not
usually consist a prominent clinical finding in early childhood with the
exemptions of Pompe and Danon disease [2] (Table II).
Children with Pompe disease can develop significant ectopy (mainly
premature ventricular contractions) or even ventricular tachycardia in
ambulatory electrocardiograms [16]. The co-existence of Danon disease
and Wolff-Parkinson-White syndrome, along with concentric hypertrophy of
left ventricle, has also been reported in literature for young patients
[36,37].
Valvular dysfunction is an additional significant
finding with the mitral valve being the most commonly affected valve.
Cases of valvular defects in childhood (from infancy to adolescence)
have been identified in literature and are related to Pompe disease,
Gaucher disease and mucopolysaccharidoses [38,39]. According to Bigg,
et al. [40], mitral valve disease in mucopoly-saccharidosis can be
associated with upregulation of enzymes (that degrade collagen or
collagen-associated proteins), as well as with accumulation of
glycosamino-glycans (that compete with proteoglycans to bind with
collagen). Macrophage infiltration seems to be the cause of mitral valve
pathology in mucopolysaccharidosis VI [41].
Toxic Intermediary Metabolites
The production of toxic intermediary products
secondary to enzymatic deficiencies is the dominating mechanism of
myocardial involvement in acidemias. The most frequent cardiac
complication in children with propionic acidemia is dilated
cardiomyopathy [2]. Romano, et al. [42] presented a series of
five neonates who developed dilated cardiomyopathy and were later
diagnosed with propionic acidemia. Furthermore, acute onset of dilated
cardiomyopathy has been reported as the only symptom of propionic
acidemia in infants and adolescents [43]. The co-existence of myocardial
involvement with methylmalonic acidemia has also been reported for both
adults and children [44]. Cardiomyopathy is of special interest in cases
of X-linked Barth syndrome (3-Methylglutaconic aciduria type II), as it
may be one of the presenting symptoms and is related with poor
prognosis. It may include hypertrophic or dilated cardiomyopathy,
although left ventricular non-compaction is the most frequent type
[17,18,45].
Isolated cases of prolonged QTc have been reported
among children with propionic acidemia, either as a presenting symptom
or as an additional finding in children already diagnosed with this
defect [19,20]. Arrhythmias have also been revealed in patients with
methylmalonic acidemia, as well as in patients with Barth syndrome
[21,46]. Few sporadic case reports in literature describe a co-existence
of congenital heart structure disorders and organic acidemias. Ebstein
cardiac anomaly and functional pulmonary atresia has been reported in a
newborn with isovaleric acidemia, while coexistence of both propionic
acidemia and cyanotic congenital heart disease has been reported in
another child [47,48]. Despite the above case reports, a clear
pathophysiological association between structural heart disorders and
metabolic defects has not yet been identified.
Clinical Features
Metabolic disorders have varying and overlapping
clinical picture [49,50]. Symptoms and signs from the cardiovascular
system are often non-specific and include shortness of breath,
hepatomegaly, edema, pathologic murmurs, failure to thrive, heart
failure and even sudden death [2]. The aforementioned symptomatology is
related to a variety of cardiac diseases (cardiomyopathy, heart rhythm
disorders, valvular dysfunction), as described above (Fig. 1).
It is noteworthy that in some cases cardiac symptoms may arise after
specific precipitating factors, such as stress, febrile illness,
fasting, dietary change and intensive exercise [50].
Diagnostic Approach
The wide variety of multisystemic presentation of
inborn errors of metabolism constitutes a diagnostic challenge for most
physicians. A systematic approach is required for the early detection of
these entities, primarily guided by a detailed medical and family
history and physical examination [2,49]. In most cases, characteristic
biochemical findings are observed, such as metabolic acidosis,
hypoglycemia, elevated creatine phospho-kinase, lactate or ammonia.
However, the definite diagnosis usually requires a more specialized
work-up based on advanced laboratory techniques. These include
assessment of plasma amino acids and acyl caritines, urine organic acids
profile, carnitine analysis, enzymatic assays or even molecular testing
[49]. The knowledge of the genetic background at an early age allows an
individualized approach to each patient, according to predicted clinical
phenotype, and promotes genetic counseling of patients and their
families [2].
The diagnosis of cardiac manifestations is usually
based on electrocardiographic and echocardiographic findings. Conduction
abnormalities and heart rhythm disorders are easily diagnosed with the
electrocardiogram, whilst echocardiography is the most easily applicable
imagining tool for the diagnosis of defects of cardiac morphology.
Simple imaging techniques (e.g., X-rays) may reveal
cardiac dilatation, while cardiac magnetic resonance imaging and
endomyocardial biopsy can exclude other morbidities (e.g.,
infectious myocarditis) [2].
Until now few "genotype-phenotype correlations" have
been described with regards to heart disorders due to inborn metabolic
errors. The deeper understanding of the genotype-phenotype correlation
provides the opportunity for more appropriate therapeutic interventions
and allows better understanding of disease expression [25].
Management of Cardiac Abnormalities
Significant progress has been made for the treatment
of metabolic diseases, especially during the last decade. A large number
of studies are still being conducted aiming for better and more targeted
therapies. Early diagnosis is crucial for the initiation of early
treatment in these patients.
The treatment approaches for inborn errors of
metabolism can be divided in two main topics: acute and chronic
treatment. The same strategy is followed for cardiac abnormalities;
treatment of acute complications and long-term management. The emergency
treatment is very important for preventing morbidity and mortality and
has to be planned even when the diagnosis is suspected. Treatment of
acute complications is based on conventional drugs (inotropes,
diuretics, antiarrhythmic drugs) and supportive measures [2].
It is important; however, to note that cardiac
complications are resistant to conventional therapies in some metabolic
defects. More specifically, cardiac function responds poorly to
treatment with diuretics and inotropes in patients with primary
carnitine deficiency. On the contrary, continued therapy with oral
L-carnitine supplements can alter the natural course of the disease and
efficiently alleviate the signs of cardiomyopathy [22]. Positive
outcomes have also been reported about the effect of carnitine
administration on heart rhythm disorders in these patients [10,11].
Long-term management strongly depends on the
underlying pathophysiology. New enzyme replacement therapies seem to
exert a beneficial effect on cardiac symptoms of patients with specific
storage disorders (e.g., Pompe and Anderson-Fabry disease)
[13,14] (Web Table I) Furthermore, liver transplantation
represents definite and curative intervention for some metabolic errors,
such as organic acidemias. In these cases, cardiomyopathy too may
reverse totally after liver transplantation [42].
Conclusions
Heart disorders are increasingly being recognized as
comorbidity in children with inborn errors of metabolism. Although there
is a lack of systematic prospective studies on this topic, the potential
adverse effect of cardiac disorders on the natural course of metabolic
defects cannot be overlooked. At a clinical level, children with
metabolic diseases should be systematically screened for cardiac
involvement during their follow-up. Furthermore, the recognition of
heart disease (especially cardiomyopathy and heart rhythm disorders) in
young patients may indicate a possible underlying metabolic defect and
promote appropriate diagnostic work-up. The correlation of cardiac
complications with specific mutations will also permit the genetic
counseling of patients and their families. On the other hand,
associating specific cardiac manifestations with specific inborn errors
of metabolism can narrow the spectrum of differential diagnosis and
contribute to a more cost-effective investigation.
Acknowledgements: Dr Panagiota Kourkoveli, Former
Locum Consultant in Heart Failure and Transplantation, Harefield
Hospital, Royal Brompton and Harefield NHS Trust, London, UK for her
valuable contribution to language editing of our manuscript.
Contributors: KP and MG participated in
literature search and drafting of the manuscript. AE had substantial
contributions to the conception of the article and supervised drafting
of the manuscript.
Funding: None; Competing interests: None
stated.
References
1. Applegarth DA, Toone JR, Lowry RB. Incidence of
inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics.
2000;105:e10.
2. Wicks EC, Elliott PM. Genetics and metabolic
cardiomyopathies. Herz. 2012;37:598-610.
3. Evangeliou A, Papadopoulou-Legbelou K, Daphnis E,
Ganotakis E, Vavouranakis I, Michailidou H, et al. Cardiac
manifestations of inborn errors of metabolism. Minerva Pediatr.
2007;59:215-8.
4. Gehrmann J, Sohlbach K, Linnebank M, Böhles HJ,
Buderus S, Kehl HG, et al. Cardiomyopathy in congenital disorders
of glycosylation. Cardiol Young. 2003;13: 345-51.
5. Leal GN, de Paula AC, Leone C, Kim CA.
Echocardiographic study of paediatric patients with
mucopolysaccharidosis. Cardiol Young. 2010;20:254-61.
6. El-Hattab AW. Inborn Errors of Metabolism. Clin
Perinatol. 2015;42:413-39.
7. Waisbren SE. Expanded newborn screening:
information and resources for the family physician. Am Fam Physician.
2008;77:987-94.
8. Xiong D, He H, James J, Tokunaga C, Powers C,
Huang Y, et al. Cardiac-specific VLCAD deficiency induces dilated
cardiomyopathy and cold intolerance. Am J Physiol Heart Circ Physiol.
2014;306:326-38.
9. Gélinas R, Thompson-Legault J, Bouchard B,
Daneault C, Mansour A, Gillis MA, et al. Prolonged QT interval
and lipid alterations beyond â-oxidation in very long-chain acyl-CoA
dehydrogenase null mouse hearts. Am J Physiol Heart Circ Physiol.
2011;301:813-23.
10. Rijlaarsdam RS, van Spronsen FJ, Bink-Boelkens
MT, Reijngoud DJ, Wanders RJ, Niezen-Koning KE, et al.
Ventricular fibrillation without overt cardiomyopathy as first
presentation of organic cation transporter 2-deficiency in adolescence.
Pacing Clin Electrophysiol. 2004;27:675-6.
11. De Biase I, Champaigne NL, Schroer R, Pollard LM,
Longo N, Wood T. Primary carnitine deficiency presents atypically with
long QT syndrome: A case report. JIMD Rep. 2012;2:87-90.
12. Wang SB, Weng WC, Lee NC, Hwu WL, Fan PC, Lee WT.
Mutation of mitochondrial DNA G13513A presenting with Leigh syndrome,
Wolff-Parkinson-White syndrome and cardiomyopathy. Pediatr Neonatol.
2008;49:145-9.
13. Winkel LP, Hagemans ML, van Doorn PA, Loonen MC,
Hop WJ, Reuser AJ, et al. The natural course of non-classic
Pompe’s disease; A review of 225 published cases. J Neurol.
2005;252:875-84.
14. Hughes DA, Elliott PM, Shah J, Zuckerman J, Coghlan
G, Brookes J, Mehta AB. Effects of enzyme replacement therapy on the
cardiomyopathy of Anderson-Fabry disease: A randomised, double-blind,
placebo-controlled clinical trial of agalsidase alfa. Heart.
2008;94:153-8.
15. Mogahed EA, Girgis MY, Sobhy R, Elhabashy H,
Abdelaziz OM, El-Karaksy H. Skeletal and cardiac muscle involvement in
children with glycogen storage disease type III. Eur J Pediatr.
2015;174:1545-8.
16. Cook AL, Kishnani PS, Carboni MP, Kanter RJ, Chen
YT, Ansong AK, et al. Ambulatory electrocardiogram analysis in
infants treated with recombinant human acid alpha-glucosidase enzyme
replacement therapy for Pompe disease. Genet Med. 2006;8:313-7.
17. Rigaud C, Lebre AS, Touraine R, Beaupain B,
Ottolenghi C, Chabli A, et al. Natural history of Barth syndrome:
a national cohort study of 22 patients. Orphanet J Rare Dis. 2013;8:70.
18. Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan
SD, Thompson WR, et al. Cardiac and clinical phenotype in Barth
syndrome. Pediatrics. 2006;118:337-46.
19. Baumgartner D, Scholl-Bürgi S, Sass JO, Sperl W,
Schweigmann U, Stein JI, et al. Prolonged QTc intervals and
decreased left ventricular contractility in patients with propionic
acidemia. J Pediatr. 2007;150:192-7.
20. Jameson E, Walter J. Cardiac arrest secondary to
long QT(C)in a child with propionic acidemia. Pediatr Cardiol.
2008;29:969-70.
21. Chao PW, Chang WK, Lai IW, Liu C, Chan KH, Tsao
CM. Acute life-threatening arrhythmias caused by severe hyperkalemia
after induction of anesthesia in an infant with methylmalonic acidemia.
J Chin Med Assoc. 2012;75:243-5.
22. Fu LJ, Chen SB, Han LS, Guo Y, Zhao PJ, Zhu M, et
al. Clinical presentation and therapeutic outcomes of carnitine
deficiency-induced cardiomyopathy. Zhonghua Er Ke Za Zhi.
2012;50:929-34.
23. Wang SM, Hou JW, Lin JL. A retrospective
epidemiological and etiological study of metabolic disorders in children
with cardiomyopathies. Acta Paediatr Taiwan. 2006;47:83-7.
24. Fu L, Huang M, Chen S. Primary carnitine
deficiency and cardiomyopathy. Korean Circ J. 2013;43:785-92.
25. Papadopoulou-Legbelou K, Gogou M, Dokousli V,
Eboriadou M, Evangeliou A.Dilated cardiomyopathy as the only clinical
manifestation of carnitinetransporter deficiency. Indian J Pediatr.
2017;84:231-3.
26. Brisca G, Fiorillo C, Nesti C, Trucco F, Derchi
M, Andaloro A, et al. Early onset cardiomyopathy associated with
the mitochondrial tRNALeu((UUR)) 3271T>C MELAS mutation. Biochem
Biophys Res Commun. 2015;458:601-4.
27. Rudaks LI, Andersen C, Khong TY, Kelly A, Fietz
M, Barnett CP. Hypertrophic cardiomyopathy with cardiac rupture and
tamponade caused by congenital disorder of glycosylation type Ia.
Pediatr Cardiol. 2012;33:827-30.
28. Kapusta L, Zucker N, Frenckel G, Medalion B, Ben
Gal T, Birk E, et al. From discrete dilated cardiomyopathy to
successful cardiac transplantation in congenital disorders of
glycosylation due to dolichol kinase deficiency (DK1-CDG). Heart Fail
Rev. 2013;18:187-96.
29. Wiles JR, Leslie N, Knilans TK, Akinbi H.
Prolonged QTc interval in association with medium-chain acyl-coenzyme A
dehydrogenase deficiency. Pediatrics. 2014;133:1781-6.
30. Yusuf K, Jirapradittha J, Amin HJ, Yu W, Hasan
SU. Neonatal ventricular tachyarrhythmias in medium chain acyl-CoA
dehydrogenase deficiency. Neonatology. 2010;98:260-4.
31. Roussel J, Labarthe F, Thireau J, Ferro F, Farah
C, Roy J, et al. Carnitine deficiency induces a short QT
syndrome. Heart Rhythm. 2016;13:165-74.
32. Trivellato M, De Palo E, Gatti R, Parenti A,
Piazza M. Carnitine deficiency as the possible etiology of idiopathic
mitral valve prolapse: Case study with speculative annotation. Tex Heart
Inst J. 1984;11:370-6.
33. Matsushita T, Sano T, Nakano S, Matsuda H, Okada
S. Successful mitral valve replacement for MELAS. Pediatr Neurol.
1993;9:391-3.
34. Shinde S, Kumar P, Mishra K, Patil N. Defect in
mitochondrial functions in damaged human mitral valve. Indian J Clin
Biochem. 2006;21:156-60.
35. Kishnani PS, Howell RR. Pompe disease in infants
and children. J Pediatr. 2004;144:35-43.
36. Cheng Z, Fang Q. Wolff-Parkinson-White syndrome
and concentric left ventricular hypertrophy in a teenager: Danon
disease. J Am Coll Cardiol. 2012;59:e7.
37. Cheng Z, Fang Q. Danon disease: focusing on
heart. J Hum Genet. 2012;57:407-10.
38. Celik S, Erdol C, Baykan M, Gokce M, Orem C,
Durmus I. Mitral valve prolapse and mitral insufficiency in two siblings
with Gaucher’s disease. Images Paediatr Cardiol. 2000;2:31-4.
39. Cripe LH, Ware SM, Hinton RB. Replacement of the
aortic valve in a patient with mucolipidosis III. Cardiol Young.
2009;19:641-3.
40. Bigg PW, Baldo G, Sleeper MM, O’Donnell PA, Bai
H, Rokkam VR, et al. Pathogenesis of mitral valve disease in
mucopolysaccharidosis VII dogs. Mol Genet Metab. 2013;110:319-28.
41. Brands M, Roelants J, de Krijger R, Bogers A,
Reuser A, van der Ploeg A, et al. Macrophage involvement in
mitral valve pathology in mucopolysaccharidosis type VI (Maroteaux-Lamy
syndrome). Am J Med Genet A. 2013;161:2550-3.
42. Romano S, Valayannopoulos V, Touati G, Jais JP,
Rabier D, de Keyzer Y, et al. Cardiomyopathies in propionic
aciduria are reversible after liver transplantation. J Pediatr.
2010;156:128-34.
43. Lee TM, Addonizio LJ, Barshop BA, Chung WK.
Unusual presentation of propionic acidaemia as isolated cardiomyopathy.
J Inherit Metab Dis. 2009;32:97-101.
44. Prada CE, Al Jasmi F, Kirk EP, Hopp M, Jones O,
Leslie ND, et al. Cardiac disease in methylmalonic acidemia. J
Pediatr. 2011;159:862-4.
45. Hanke SP, Gardner AB, Lombardi JP, Manning PB,
Nelson DP, Towbin JA, et al. Left ventricular noncompaction
cardiomyopathy in Barth syndrome: An example of an undulating cardiac
phenotype necessitating mechanical circulatory support as a bridge to
transplantation. Pediatr Cardiol. 2012;33:1430-4.
46. Spencer CT, Byrne BJ, Gewitz MH, Wechsler SB, Kao
AC, Gerstenfeld EP, et al. Ventricular arrhythmia in the X-linked
cardiomyopathy Barth syndrome. Pediatr Cardiol. 2005;26:632-7.
47. Qadi AM, Hamadah HK, Jijeh AM, Hijazi OM, Kabbani
MS. Ebstein cardiac anomaly, functional pulmonary atresia and isovaleric
acidemia: A case report. J Saudi Heart Assoc. 2014;26:170-3.
48. Palermo RA, Monge MC, Charrow J, Costello JM,
Epting CL. Masquerading acidosis after cardiopulmonary bypass: A case of
propionic acidemia and congenital heart disease. World J Pediatr
Congenit Heart Surg. 2015;6:291-4.
49. Burton BK. Inborn errors of metabolism in
infancy: a guide to diagnosis. Pediatrics. 1998;102:E69.
50. Ahrens-Nicklas RC, Slap G, Ficicioglu C.
Adolescent presentations of inborn errors of metabolism. J Adolesc
Health. 2015;56:477-82.


