|
|
|
Indian Pediatr 2016;53: 738-740 |
 |
Isoleucine Deficiency in
a Neonate Treated for Maple Syrup Urine Disease Masquerading as
Acrodermatitis Enteropathica
|
|
Benjamin Ross, Manish Kumar,
*Hema Srinivasan and
#Alka V Ekbote
From Departments of Neonatology, *Pediatrics and
#Clinical Genetics Unit,Christian
Medical College, Vellore, Tamilnadu, India.
Correspondence to: Dr Benjamin Ross, Department of
Neonatology, Christian Medical College, Vellore 632 004, Tamilnadu,
India.
Email:
[email protected]
Received: June 16, 2015;
Initial review: August 20, 2015;
Accepted: April 19, 2016.
|
Background: Special diet with restricted branched-chain-amino-acids
used for treating maple syrup urine disease can lead to specific amino
acid deficiencies. Case characteristics: We report a neonate who
developed skin lesions due to isoleucine deficiency while using
specialised formula. Intervention/Outcome: Feeds were
supplemented with expressed breast milk. This caused biochemical and
clinical improvement with resolution of skin lesions. Message:
Breast milk is a valuable and necessary adjunct to specialized formula
in maple syrup urine disease to prevent specific amino acid deficiency
in the neonatal period.
Keywords: Branched-chain amino acid, Restrictive diets,
Treatment.
|
|
Maple Syrup Urine Disease (MSUD) is an autosomal
recessive inherited inborn error of metabolism, which can be managed by
specific dietary modifications to reduce branched chain amino acid
intake [1]. If not treated early, irritability and poor feeding can
manifest within 2-3 days of life and encephalopathy with opisthotonus
and intermittent apnea can start by day 4-5 [2]. Coma and respiratory
failure occur by 7-10 days [2].
We report a baby with antenatally diagnosed MSUD who
developed isoleucine deficiency while on specialised formula in the
neonatal period and presented with cutaneous lesions mimicking
acrodermatitis enteropathica. The skin lesions resolved after addition
of breast milk to the diet with normalization of isoleucine levels.
Case Report
A 29-year-old gravida 2, para 1 lady in a second
degree consanguineous marriage presented to the clinic for prenatal
testing. Her first baby had seizures in the neonatal period and was
later diagnosed to have MSUD. He had global developmental delay with
seizures and died at 2 years of age.
During this pregnancy, both parents were counseled
about the chances of recurrence of MSUD in the baby. Sanger sequencing
of branched chain keto acid dehydrogenase E1 alpha polypeptide (BCKDHA)
and branched chain ketoacid dehydrogenase E1 beta polypeptide (BCKDHB)
genes were done on the couple and chorionic villous sample from the
fetus. All the variants were analyzed and annotated using Human
(GRCh38.p3) public databases using Ensembl and HGMD (Human Gene Mutation
Database). In silico analysis was performed for all the
variants using SIFT (Sorting Intolerant From Tolerant) and POLYPHEN2
(Polymorphism Phenotyping version 2).
Both parents were detected to be heterozygous for the
BCKDHA gene mutation (c.757 G>A; p.Ala253Ser.). Mother was
heterozygous for BCKDHB variation (c.1039-5T>C.). Father did not
have any variation of the BCKDHB gene. The BCKDHB
variation is a novel variant of unknown significance and is not known to
cause MSUD. Chorionic villous sample was homozygous for the c.757 G>A;
p.Ala253Ser. mutation in the BCKDHA gene, a known mutation
causing MSUD. Based on these results, a diagnosis of MSUD was made in
the fetus.
Both parents were extensively counseled about the
nature of the disease before and after the results. However, they opted
to continue the pregnancy. The neonate was delivered uneventfully by
caesarian section at 39 weeks gestation. Due to a delay in shipment of
specialised formula for MSUD from the USA, the neonate was started on
intravenous fluids. MSUD-specific formula was started on day 5. Initial
biochemical analysis before starting the formula showed a valine level
of 423µmol/L (Normal 80-246 µmol/L), leucine level of 217 µmol/L
(Normal: 46-109 µmol/L) and isoleucine level of 153 µmol/l (Normal:
27-53 µmol/L). In order to avoid encephalopathy and neurological
sequelae the neonate was not started on breast milk.
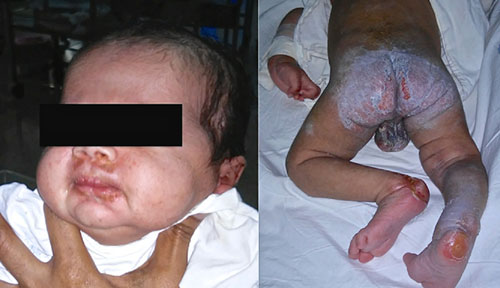 |
|
Fig. 1 Skin lesions in the perioral,
perianal regions and feet mimicking acrodermatitis enteropathica
seen in an infant with isoleucine deficiency.
|
On day 8, the neonate developed watery stools. On day
10, the neonate developed excoriative skin lesions in the perianal and
perioral regions, and both feet which rapidly progressed (Fig.
1); scalp was spared and hair was normal. On day 12, the neonate
developed fever and lethargy. The neonate was started on intravenous
antibiotics (cloxacillin, cefaperazone and amikacin). Blood culture
revealed Klebsiella sp. sensitive to cefaperazone. The
neonate became afebrile, active and well within 48 hours of starting
antibiotics. However, the skin lesions continued to worsen. An amino
acid profile revealed Isoleucine level 8 µmol/L, Leucine level 232
µmol/L and Valine level 226 µmol/L. Zinc deficiency causing
acrodermatitis enteropathica was ruled out as serum zinc levels were
normal (74 µg/dL). Staphylococcal sepsis was ruled out as blood culture
and skin swab culture did not show growth of S. aureus. A final
diagnosis of acrodermatitis enteropathica like skin lesions due to low
isoleucine levels was made based on the clinical profile and isoleucine
level profiles through this time period. Expressed breast milk was added
to the feeds to amount to 30% of total feed volume to compensate for
isoleucine deficiency. This amount was arbitrarily chosen with a plan to
modify it based on further clinical and biochemical progress. The
neonate showed a rapid improvement in the skin lesions which started
healing. Repeat amino acid profile showed a normal isoleucine levels (66
µmol/L).
Discussion
Management of MSUD involves use of specialized diet
with restricted branched chain amino acids [1]. However, amino acid
profile needs to be checked frequently to ensure that branched chain
amino acid levels are within normal levels. Elevated levels of branched
chain amino acids can result in encephalopathy, seizures and
developmental delay [1].
The neonate, described herein, developed isoleucine
deficiency and presented with skin lesions similar to acrodermatitis
enteropathica. These skin lesions have also been described during
treatment of organic acidemias (methyl malonic acidemia, glutaric
aciduria and propionic acidemia) [3,4] and amino-acidopathies (maple
syrup urine disease) [5,8] due to iatrogenic deficiency of isoleucine.
It has been called acrodermatitis acidemica [3-9] or acrodermatitis
dysmetabolica [9]. Similar lesions have also been described in biotin
deficiency and free fatty acid deficiency [10]. The neonate described
here had the clinical profile and response to treatment similar to the
cases with isoleucine deficiency described previously.
Special formulae with restricted branched chain amino
acids is essential for managing neonates with MSUD. However, when used
alone even for a short time, neonates can quickly develop branched chain
amino acid deficiency as highlighted in this case report. Expressed
breast milk (EBM) supplementation is an effective way to avoid and treat
deficiency of branched chain amino acid in the neonatal period and early
infancy in these babies as was seen in the neonate described herein. The
amount of EBM to be added needs to be adjusted depending on the clinical
response and repeated amino acid profile testing. The other options to
prevent deficiency of branched chain amino acids include supplementation
with specific amino acids, use of infant formula and low dose protein
supplementation. It is also necessary to frequently monitor amino acid
profiles in the growing infants to optimize their dietary intake, growth
and ensure normal levels of branched chain amino acids.
Contributors: BR: carried out the literature
review and drafted the manuscript; BR, MK and HS: were responsible for
the diagnosis and management; AE: was involved in antenatal genetic
diagnosis and prenatal counselling.
Funding: None; Competing interests: None
stated.
References
1. Gleason J. Avery’s Diseases of the Newborn. 9th
ed. Philadelphia, PA: Saunders; 2011. p. 221-3.
2. GeneReviews. Strauss KA, Puffenberger EG, Morton
DH. Maple Syrup Urine Disease. Available from:
http://www.ncbi.nlm.nih.gov/books/NBK1319/. Accessed June 15, 2015.
3. Niiyama S, Koelker S, Degen I, Hoffmann GF, Happle
R, Hoffmann R. Acrodermatitis acidemica secondary to malnutrition in
glutaric aciduria type I. Eur J Dermatol. 2001;11:244-6.
4. De Raeve L, De Meirleir L, Ramet J, Vandenplas Y,
Gerlo E. Acrodermatitis enteropathica-like cutaneous lesions in organic
aciduria. J Pediatr. 1994;124:416-20.
5. Templier I, Reymond JL, Nguyen MA, Boujet C,
Lantuejoul S, Beani JC, et al. Acrodermatitis enteropathica-like
syndrome secondary to branched-chain amino acid deficiency during
treatment of maple syrup urine disease. Ann Dermatol Venereol.
2006;133:375-9.
6. Bosch AM, Sillevis Smitt JH, Van Gennip AH,
Abeling NG, Schutgens RB, Bakker HD, et al. Iatrogenic isolated
isoleucine deficiency as the cause of an acrodermatitis enteropathica-like
syndrome. Br J Dermatol. 1998;139:488-91.
7. Domínguez-Cruz JJ, Bueno-Delgado M, Pereyra J,
Bernabeu-Wittel J, Conejo-Mir J. Acrodermatitis enerophatica-like skin
lesions secondary to isoleucine deficiency. Eur J Dermatol.
2011;21:115-6.
8. Dominguez-Cruz J, Bueno-Delgado M, Bernabeu-Wittel
J, Delgado-Pecellin C, Conejo-Mir J. Acrodermatitis acidemica:
experience of 5 years in Andalusia. Pediatr Dermatol. 2010;27:218–9;
author’s reply 219-20.
9. Tabanlioðlu D, Ersoy-Evans S, Karaduman A.
Acrodermatitis enteropathica-like eruption in metabolic disorders:
acrodermatitis dysmetabolica is proposed as a better term. Pediatr
Dermatol. 2009;26:150-4.
10. Gehrig KA, Dinulos JGH. Acrodermatitis due to nutritional
deficiency. Curr Opin Pediatr. 2010;22: 107-12.
|
|
|
 |
|
