|
|
|
Indian Pediatr 2016;53: 695-701 |
 |
Metabolic
Liver Diseases Presenting as Acute Liver Failure in Children
|
|
Seema Alam and Bikrant Bihari Lal
From Department of Pediatric Hepatology, Institute of
Liver and Biliary Sciences, New Delhi, India.
Correspondence to: Prof Seema Alam, Professor and
Head, Department of Pediatric Hepatology, Institute of Liver and Biliary
Sciences, New Delhi 110 070, India.
Email: [email protected]
|
Context: Suspecting metabolic liver disease in an infant or
young child with acute liver failure, and a protocol-based workup for
diagnosis is the need of the hour. Evidence acquisition:
Data over the last 15 years was searched through Pubmed using the
keywords "Metabolic liver disease" and "Acute liver failure" with
emphasis on Indian perspective. Those published in English language
where full text was retrievable were included for this review.
Results: Metabolic liver diseases account for 13-43% cases of
acute liver failure in infants and young children. Etiology remains
indeterminate in very few cases of liver failure in studies where
metabolic liver diseases were recognized in large proportion.
Galactosemia, tyrosinemia and mitochondrial disorders in young children
and Wilson’s disease in older children are commonly implicated. A high
index of suspicion for metabolic liver diseases should be kept when
there is strong family history of consanguinity, recurrent abortions or
sibling deaths; and history of recurrent diarrhea, vomiting, failure to
thrive or developmental delay. Simple dietary modifications and/or
specific management can be life-saving if instituted promptly.
Conclusion: A high index of suspicion in presence of red flag
symptoms and signs, and a protocol-based approach helps in timely
diagnosis and prompt administration of life-saving therapy.
Keywords: Acute liver failure, Etiology, Infants, Liver
Transplantation.
|
|
Inborn errors of metabolism, where hepatomegaly
and/or abnormal liver functions form part of the clinical disease, are
collectively referred to as Metabolic liver diseases (MLD). MLD can have
varied presentations in infants and children, most common of them being:
(i) organomegaly, (ii) encephalopathy due to
hyperammonemia and/or primary lactic acidemia, (iii) pediatric
acute liver failure (ALF), (iv) cirrhosis with or without portal
hypertension, and (v) cholestatic liver disease. A high index of
suspicion for MLD is important as urgent intervention such as dietary
manipulation or disease-specific treatment may be life-saving. The
outcome of patients undergoing liver transplantation for MLD has
improved considerably over the last decade. Moreover, it is important to
establish the correct diagnosis, so that appropriate genetic counselling
can be offered to the family. MLD merit special attention in
differential diagnosis of pediatric ALF, especially in infants and young
children in whom they constitute 13-43% of all cases (Web Table
I) [1-9].
The Pediatric ALF study group definition can be used
to define acute liver failure in infants and children [6]. The group
enlists criteria for defining ALF as follows: (i) children with
no known evidence of chronic liver disease (CLD), (ii)
biochemical evidence of acute liver injury, and (iii) hepatic-based
coagulopathy defined as International normalized ratio (INR)
³1.5 not corrected by
vitamin K in the presence of clinical hepatic encephalopathy or INR
³2 regardless
of the presence or absence of clinical hepatic encephalopathy.
Neonatal liver failure is defined as "failure of the synthetic
function of liver within 4 weeks of birth’’ [10]. Presence of
encephalopathy is not mandatory for defining acute liver failure in
infants as it is often very difficult to recognize. Moreover,
encephalopathy may be a very late event in the course of the disease
[1,5]. We have previously reported that average jaundice to
encephalopathy interval is significantly higher in pediatric ALF due to
MLD group vis-à-vis other etiologies [1]. Another important
difference is that complete absence of evidence of CLD cannot be kept as
prerequisite, especially when the etiology is a suspected MLD [4]. MLD
patients may have variable degrees of liver damage before clinical
presentation, and overt signs and stigmata of chronic liver disease may
be present.
Metabolic Causes of Acute Liver Failure
MLD are an important causes of pediatric ALF,
especially in neonates, infants and young children. MLD account for
13-43% of acute liver failure in younger children, while accounting for
only 5-20% of ALF in older children [1-9] (Web Table I).
Studies which focus on infants and young children have higher prevalence
of MLD as compared to the studies which include older children. The
proportion of cases with indeterminate etiology are higher (38-53%)[4-6]
in studies, where the proportion of MLD cases is lower (13-19%). On the
other hand, the studies with higher prevalence (33-43%) of MLD among
pediatric ALF [1,3,7] had much smaller proportion (13-18%) of cases
remaining indeterminate. Narkewicz, et al. [11] retrospectively
analyzed the workup of children labelled as indeterminate in pediatric
ALF study group, and found that 54% of these children had not been
screened for some common metabolic disorders, before assigning them as
indeterminate. In an earlier Indian study, 4 children among 67 with
fulminant liver failure were reported to be non A, non E, but MLD was
not reported [12]. However, this series included only one infant, and
the definition used for ALF was different. With advent of better
diagnostic protocols, MLDs are more frequently diagnosed now [1].
Some of the authors have earlier listed neonatal
hemochromatosis as a MLD. Being a gestation-associated alloimmune
disorder, it has now been excluded from the list of MLDs. Galactosemia,
tyrosinemia, mitochondrio-pathies and fatty acid oxidation defect (FAOD)
are the commonest metabolic diseases presenting as ALF in infants.
Wilson’s disease is the commonest MLD presenting as ALF among older
children, others being mitochondriopathies, FAOD and urea cycle defects.
Pathophysiology
In disorders such as galactosemia, tyrosinemia and
Urea cycle defects, the pathogenesis of MLD can be attributed to a
defect in the intermediary metabolic pathway leading to the accumulation
of toxic metabolites (formed in one of the preceding steps) which leads
to liver failure. These conditions present after a symptom-free interval
before clinical signs of ‘intoxication’ appear. Acute attacks may be
preceded by catabolic states, fever, intercurrent illnesses and specific
food intake. Most of these disorders are treatable and require emergency
removal of the toxin by using special diets, extracorporeal procedures,
drugs or vitamins [13]. Another pathogenetic mechanism is an energy
deficiency state. The mitochondrial energy defects encompass the
congenital lactic acidemias, respiratory chain disorders, pyruvate
oxidation defects and FAOD. Cytoplasmic energy defects include disorders
of glycolysis, glycogen metabolism, gluconeogenesis and the pentose
phosphate pathways. The metabolic defects with energy-deficient states
present early and can even have prenatal onset. In lysosomal disorders
(Wolman disease and cholesteryl ester storage disorder), peroxisomal
disorders (Zellweger syndrome) and congenital defect of glycosylation,
liver failure occurs due to involvement of cellular organelles.
Suspecting MLD as Cause of ALF
Box 1 enumerates the important points in
history that should raise the suspicion of a metabolic disorder.
Children with MLD presenting as ALF tend to be younger. A strong family
history of consanguinity, recurrent abortions, sibling deaths and
previously affected children are strong pointers to the possible
etiology of MLD. History of recurrent diarrhea and vomiting, failure to
thrive and developmental delay are other indicators suggesting MLD
[1,3]. Patients with MLD tend to have a longer jaundice to
encephalopathy interval. Neurological involvement in form of hypotonia,
myopathy, seizures, ophthalmoplegia, psychomotor dysfunction or presence
of multisystem involvement should raise the suspicion of a mitochondrial
depletion syndrome [14].
|
Box 1 Clinical Pointers of Metabolic
Liver Diseases
|
|
• Consanguinity, abortions and neonatal deaths
• SIDS and psychiatric illnesses
• Recurrence in times of catabolic stress (fever, exercise,
prolonged fasting)
• Recurrent vomiting, diarrhea, failure to thrive , short
stature, dysmorphic features, edema
• Seizures, early morning irritability, lethargy-
hypoglycemic symptoms
• Developmental delay, hypotonia and seizures, cataract,
unusual odours, rickets, and renal tubulopathy
• Jaundice, hepatosplenomegaly and hepatic failure,
hypoglycemia, lactic acidemia, hyperammonemia and coagulopathy
• Relation to specific foods like aversion to sugars in HFI,
dislike for proteins in urea cycle defects
SIDS: Sudden Infant Death Syndrome, HFI: Hereditary Fructose
Intolerance.
|
A meticulous dietary history can help guide the
clinician. Onset of liver dysfunction on milk feeds points towards
diagnosis of galactosemia, whereas onset of symptoms after introduction
of complementary foods (containing fructose or sucrose) points towards
hereditary fructose intolerance that can also present in those receiving
fructose in form of honey, syrups or formula milk containing fructose or
sorbitol. Aversion to sugars and sweet foods in older children also
suggests this disorder. Some MLD have their typical age of presentation
and age-appropriate differential diagnosis should be considered (Table
I). Encephalopathy is difficult to diagnose in children and could be
disproportionately more severe to the liver dysfunction in urea cycle
defects and primary lactic acidemias. Developmental delay,
cardiomyopathy, renal tubulopathy in a hypotonic child with convulsive
disorder with or without treatment with valproic acid could be a setting
for mitochondrial disorders.
TABLE I Categorization of Metabolic Liver Diseases by age of Presentation
|
0-6 mo |
Galactosemia, Tyrosinemia type 1, Mitochondrial cytopathy and
Wolman’s disease |
|
6 mo - 3 y |
Tyrosinemia type I, FAOD, Mitochondrial cytopathy, Galactosemia,
HFI, UCD and CDG |
|
Older children |
Wilson’s disease, FAOD, Mitochondrial Cytopathy, HFI, UCD and
CDG |
|
FAOD: Fatty acid oxidation defect, HFI: Hereditary fructose
intolerance, UCD: Urea cycle defect, CDG: Congenital disorders
of glycosylation. |
Children with MLD have much higher bilirubin, more
severe synthetic dysfunction, hypoglycemia and hyperammonemia as
compared to those with ALF not related to MLD [1,3]. Children with MLD
tend to have a much higher bilirubin, but lower transaminases, gamma-glutamyl
transferases (GGT) and INR as compared to the viral causes of ALF [4].
Approach to MLD presenting as ALF
The algorithmic diagnostic approach to an infant or
young child with ALF and suspected MLD is depicted in Fig 1.
For an older child, the etiological list includes only WD, HFI,
mitochondrial defect and FAOD. Except WD, all others have been covered
in the above mentioned algorithm. WD should be suspected in a patient
with ALF who have KF ring on slit-lamp examination, Coombs negative
hemolytic anemia, low serum uric acid levels (<2.5 mg/dL), low serum
alkaline phosphatase (SAP) activity (SAP: bilirubin ratio <4) and
increased AST:ALT ratios [15]. As per European Association for Study of
Liver (EASL) guidelines, serum ceruloplasmin <10 mg/dL is contributory
for definitive diagnosis of WD [16], but ceruloplasmin, being an acute
phase reactant, can be falsely normal in children with acute liver
failure. Hence, 24 hours urinary copper (>100 µg/day) and KF ring are
important for diagnosis of WD in setting of ALF.
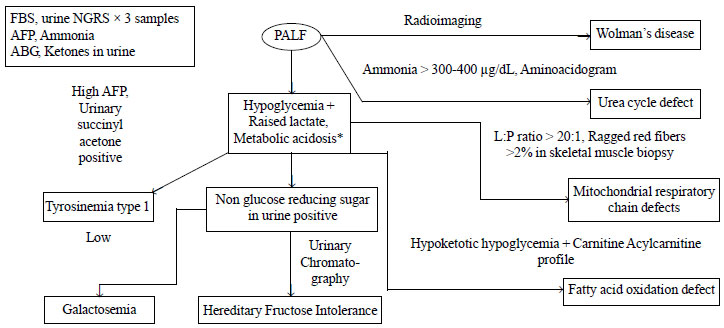 |
|
PALF: Pediatric acute liver failure, MLD: Metabolic liver
disease, FBS: Fasting blood sugar, NGRS : Non glucose reducing
sugar, AFP: Alphafeto protein , L: P = Lactate: Pyruvate, ABG:
Arterial blood gases, GALT : Galactose-1-PO4 Uridyltransferase.
*Hypoglycemia/acidosis/ raised lactate may occur in ALF with non
MLD also due to liver injury/ sepsis/ hypotension.
Fig. 1 Approach to a child with Acute
liver failure and suspected Meabolic liver disease.
|
A careful history and examination should narrow down
the differential diagnosis and establish the degree of liver
dysfunction. The time of onset of the symptoms and the rapidity of
progression can give a clue to diagnosis. First line metabolic screen
should be done in all cases of pediatric ALF, that includes three
consecutive samples of urinary non-glucose reducing substances, ketones,
arterial blood gas analysis, and serum lactate, serum alpha-feto protein
and blood ammonia levels. Non-glucose reducing substances in urine can
be identified by testing for reducing substances in urine by Benedict
test, and then demonstrating absence of glucose by dipstick method. If
suspecting galactosemia, it should be ensured that the child was on
galactose-containing diet when the urinary samples were examined. In
liver failure, blood sugar may be low and lactate may be high due to
advanced liver disease. On the basis of the metabolic screen, we can
narrow down on the possible etiology of the MLD. The specific diagnostic
tests for the common MLDs presenting as pediatric ALF is shown in
Table II. It is important to remember that galactose-1-phosphate
uridyl transferase (GALT) assay is not reliable if the child has
received blood transfusion in the preceding three months. In such cases,
it is advisable to continue the galactose free diet, till we can get
reliable results of the GALT assay.
TABLE II Confirmatory Tests and Management of Common Metabolic Liver Diseases
|
Disorder |
Confirmatory Test |
Management |
|
Galactosemia |
Galactose-1-PO4 Uridyl-transferase assay |
Galactose-free diet |
|
Hereditary Fructose |
Fructo-aldolase B assay in liver tissue * |
Fructose-free diet |
|
Intolerance |
Urine chromatography to show fructose |
|
|
Mutational analysis |
|
|
Tyrosinemia type 1 |
Urinary succinylacetone phenylalanine diet ** |
NTBC* + Low tyrosine and |
|
Urea Cycle Defect |
Plasma aminoacidogram to show the levels of citrulline |
Ammonia scavengers, protein |
|
(UCD) |
and arginine based on which the type of UCD can be |
free diet with essential amino |
|
decidedOrotic acid estimation in urine to diagnose |
acids supplementation |
|
OTC**deficiency |
|
|
Fatty Acid Oxidation |
Carnitine - acyl carnitine profile |
Avoid prolonged fasting |
|
Defect |
|
Breastfeeding in MCAD MCT rich diet in VLCAD and LCHAD |
|
|
Carnitine in carnitine transporter |
|
|
deficiency Bezafibrate* in VLCAD |
|
Respiratory chain disorder |
Analysis of oxidative phosphorylation complexes I-IV from intact
mitochondria isolated from fresh skeletal muscle* |
Normocaloric and low carbohydrate diet |
|
Oral Coenzyme Q for CoQ10 deficiency |
Avoidance of certain drugs |
|
|
Carnitine in carnitine deficiency |
| |
|
|
|
Wilson Disease |
24 hour urine copper |
Chelation therapy with D- |
|
KF ring Serum CeruloplasminSAP:Bilirubin ratio < 4 |
Penicillamine started at 10 mg/kg/d |
|
AST/ALT ratio > 4 |
and increased to 20 mg/kg/d, Zinc & Pyridoxine |
|
NTBC: 2-nitro-4-trifluoro-benzoyl-cyclohexane-1,3-dione,
OTC: Ornithine transcarbamylase, MCAD: Medium chain Acyl-CoA
Dehydrogenase, MCT: Medium Chain Triglyceride, VLCAD: Very long
chain Acyl-CoA Dehydrogenase, LCHAD: Long chain
3-hydroxyacyl-CoA Dehydrogenase, CoQ10: Coenzyme Q 10, SAP:
Serum alkaline phosphatase, AST: Aspartate aminotransferase,
ALT: Alanine aminotransferase, KF ring: Kayser Fleischer ring.
*These tests and therapeutic options are not available in India;
**OTC samples are being sent outside the country by Indian
laboratories; *Low Tyrosine and Phenylalanine diet is being
marketed in India but not manufactured. |
Management
With improvement in supportive management, the
outcome of pediatric ALF has improved considerably. Liver
transplantation may be life-saving for children who fail to respond to
conservative management. Whenever MLD is a strong possibility, all feeds
should be withdrawn for 24-48 hours awaiting first line investi-gations.
The dietary modifications can be modelled on the basis of differentials
considered based on history and examination. Appropriate feeds can be
introduced based on final diagnosis. The main aim of withholding feeds
is to stop further accumulation of potentially toxic metabolites, but at
the same time further catabolic breakdown of body stores should be
avoided as it can worsen liver failure. Intravenous infusion of 10%
dextrose with required electrolytes is appropriate for most cases. The
exception is congenital lactic acidosis and mitochondrial disorders
where a 5% dextrose-based solution should be used as high carbohydrate
supply may exacerbate the lactic acidosis. Restriction of protein to 0.5
-1 g/kg/day (half in form of essential amino acids) is recommended for
management of urea cycle defects. If FAOD has been excluded, then
intralipid should be added at 1g/kg/day to boost energy intake.
Supportive management for pediatric ALF includes
glucose for maintaining normoglycemia, correction of coagulopathy in
case of bleeding, antibiotics for sepsis, and maintenance of fluid and
electrolyte balance. Although most data is from cirrhotics, but among
anti-ammonia measures, polyethylene glycol 3350 was more effective than
lactulose with resolution of encephalopathy in 90% and 50% cases,
respectively [17]. A systemic review states that there is insufficient
evidence to support or refute the use of non-absorbable disaccharides (Lactulose
and Lactitol) for hepatic encephalopathy. Antibiotics (Rifaximin and
Neomycin) were superior to non-absorbable disaccharides in improving
encephalo-pathy, but it is unclear whether this difference is clinically
important [18]. Algorithmic approach of management of hepatic
encephalopathy also mentions usage of lactulose and rifaximin [19]. In a
report from US ALF study group, lactulose increased survival time but
had no effect on overall outcome [20]. Sodium benzoate has been
encouraged keeping in view it is as effective as lactulose but 10 times
less expensive [21]. Sodium Benzoate is recommended for use in HE due to
urea cycle defects [22]. Definitive management for common MLD is
depicted in Table II.
Liver Assist Devices
The role of the support device (artificial or
bio-artificial) in ALF has an objective to either support the patient
until the native liver recovers, or to bridge the patient to liver
transplantation. Artificial support therapies (plasma exchange,
hemodialysis and Molecular Adsorbents Recirculating System) provide
detoxification support without the use of cellular material. Molecular
Adsorbents Recirculating System has been reported to be more beneficial
than combined plasma exchange and hemodialysis [24]. In another study,
it was found to be beneficial in decreasing ammonia levels in
adolescents but there were no benefits in infants in whom the device was
poorly tolerated [25]. There is scarcity of data regarding the role of
liver assist devices in management of MLD. Intermittent and continuous
hemodialysis are effective modalities for the acute management of urea
cycle defects and organic acidemia [26]. The pre-procedure physiological
condition of the patient is the main determinant of outcome [27].
Moreover, most of the artificial liver assist devices help during
hepatic encephalopathy but do not improve overall survival in ALF.
Bio-artificial systems use cellular material to provide detoxification
and liver’s synthetic functions. A variety of such systems have been
tested in non-randomized trials, but are not recommended outside
clinical trials.
Liver Transplantation
The advent of successful liver transplantation has
revolutionized management of children with MLD who fail to respond to
conservative management. Galactosemia, HFI, tyrosinemia type 1 and urea
cycle defects may not respond to medical therapy and dietary
restrictions if diagnosed late, and in an emergency liver
transplantation may prove to be life-saving. Although individually rare,
when considered together, MLD represents approximately 15-25% of
indications for pediatric liver transplantation [28]. MLD are the second
most common indication for liver transplantation after biliary atresia
[29]. UCD, alpha-1-antitrypsin deficiency, cystic fibrosis, WD and
tyrosinemia type 1 are the common MLD requiring liver transplantation in
children. Post-transplant survival for children with MLD is comparable
to those with other diseases with a better graft survival than those
with other diseases [29]. A better outcome of liver transplantation in
MLD could be attributed to the fact that many children with MLD
underwent liver transplantation to correct an enzymatic defect, and did
not have structural (parenchymal) liver disease.
Liver transplantation has been successfully done in
many cases of tyrosinemia, galactosemia, mitochondrio-pathies and UCD
presenting as ALF [28-30]. Liver transplantation is usually
contraindicated in diseases with severe multisystemic involvement
e.g. mitochondrial defect with severe neurological involvement/cardiomyopathy.
A rapid assessment of the severity of extrahepatic involvement in a
child with mitochondrio-pathy and decompensating liver is mandatory, so
as to take a decision about the usefulness of liver transplantation in
such a case. Suitability of heterozygous parents as donors is another
important issue to be resolved.
Although Wilson disease presenting with
encephalo-pathy is invariably fatal and can be treated only by liver
transplantation, the decision to list a child with this disorder without
encephalopathy is very difficult [5,31]. Revised King’s score for this
disorder [31] had been previously shown to be efficacious in predicting
the survival with native liver [31,32]. However, doubts have been raised
recently over the ability of this score to predict mortality without
liver transplantation [33]. Survival is difficult to predict and
continued investigations for predictors of outcome in Wilson disease are
necessary.
Hepatocyte transplantation is moderately successful
for MLD presenting as ALF, as a bridge to liver transplantation [34].
Hepatocyte transplantation holds promise as an alternative to organ
transplantation and numerous animal studies indicate that transplants of
isolated liver cells can correct metabolic deficiencies of the liver.
Stem cell based technology is a new biotechnology approach to treat
patients with MLD. Adult liver stem cells can differentiate into
hepatocyte like cells and can be infused in the recipient’s liver to
activate a missing metabolic function. The percentage of liver cell
replacement considered as necessary to significantly improve metabolic
disorders is around 5% of the total liver mass, while 10% could
normalize the function [35,36].
Genetic Counseling
Parents who have a child with MLD must undergo
genetic couseling. The probability of the next sibling being affected
from the disease should be explained, and prenatal testing and
counseling should be offered where available. The parents must be
explained about the nature of the illness and risk of occurrence in
future pregnancies. Prenatal diagnosis of tyrosinemia is possible by
analysis of succinylacetone in amniotic fluid supernatant and by assay
of fumaryl acetoacetate hydrolase in cultured amniotic fluid cells or
chorionic villus material [37]. Similarly, a GALT assay can be planned
early for the next child of parents who already have a child suffering
from galactosemia.
Conclusion
Metabolic liver diseases account for 13-43% of cases
of ALF in infants and young children. Many of these conditions are
potentially curable with dietary modifications or medications if
recognized early. A high index of suspicion in presence of red flag
symptoms and signs is need of the hour. A protocol-based approach will
identify the etiology in most of the patients. Liver transplantation has
markedly improved the outcome of MLD in children.
Contributors: Both authors conceptualized
the work, searched and reviewed the data. BBL prepared the first draft;
SA: critically reviewed and revised the manuscript. Both authors
approved the final version.
Funding: None; Competing interests: None
stated.
References
1. Alam S, Lal BB, Khanna R, Sood V, Rawat D. Acute
liver failure in infants and young children in a specialized pediatric
liver centre in India. Indian J Pediatr. 2015 Jan 6. [Epub ahead of
print].
2. Rajanayagam J, Coman D, Cartwright D, Lewindon PJ.
Pediatric acute liver failure: Etiology, outcomes, and the role of
serial pediatric end-stage liver disease scores. Pediatr Transplant.
2013;17:362-8.
3. Brett A, Pinto C, Carvalho L, Garcia P, Diogo L, Gonçalves
I. Acute liver failure in under two year-olds–are there markers of
metabolic disease on admission? Ann Hepatol. 2013;12:791-6.
4. Sundaram SS, Alonso E, Narkewicz MR, Zhang S,
Squires RH and Pediatric Acute Liver Failure Study Group.
Characterization and outcome of young infants with acute liver failure.
J Pediatr. 2011;159:813-8.
5. Dhawan A. Etiology and prognosis of acute liver
failure in children. Liver Transplant. 2008;14:S80-S4.
6. Squires RH, Shneider BL, Bucuvalas J, Alonso E,
Sokol RJ, Narkewicz MR, et al. Acute liver failure in children:
The first 348 patients in pediatric acute liver failure study group. J
Pediatr. 2006;148:652-8.
7. Durand P, Debray D, Mandel R, Baujard C,
Branchereau S, Gauthier F, et al. Acute liver failure in infancy:
A 14-year experience of a pediatric liver transplantation center. J
Pediatr. 2001;139:871-6.
8. Kaur S, Kumar P, Kumar V, Sarin SK, Kumar A.
Etiology and prognostic factors of acute liver failure in children.
Indian Pediatr. 2013;50:677-9.
9. Lee WS, McKiernan P, Kelly DA. Etiology, outcome
and prognostic indicators of childhood fulminant hepatic failure in the
United Kingdom. J Pediatr Gastroenterol Nutr. 2005;40:575-81.
10. Shanmugam NP, Bansal S, Greenough A, Verma A,
Dhawan A. Neonatal liver failure- etiologies and management- state of
the art. Eur J Pediatr. 2011;170: 573-81.
11. Narkewicz MR, DellOlio D, Karpen SJ, Murray KF,
Schwarz K, Yazigi N, et al. Pattern of diagnostic evaluation for
the causes of pediatric acute liver failure: an opportunity for quality
improvement. J Pediatr. 2009;155:801-6.
12. Poddar U, Thapa BR, Prasad A, Sharma AK, Singh K.
Natural history and risk factors in fulminant hepatic failure.
Arch Dis Child. 2002;87:54-6.
13. Boles RG, Buck EA, Blitzer MG, Platt MS, Cowan
TM, Martin SK, et al. Retrospective biochemical screening
of fatty acid oxidation disorders in postmortem livers of 418 cases of
sudden death in the first year of life. J Pediatr. 1998;132:924-33.
14. Dimmock DP, Zhang Q, Dionisi-Vici C, Carrozzo R,
Shieh J, Tang LY, et al. Clinical and molecular features of
mitochondrial DNA depletion due to mutations in deoxyguanosine kinase.
Hum Mutat. 2008;29:330-1.
15. Korman JD, Volenberg I, Balko J, Webster J,
Schiodt FV, Squires RH, et al. Screening for Wilson disease in
acute liver failure: a comparison of currently available diagnostic
tests. Hepatology. 2008;48:1167-74.
16. Eurpean Association for Study of Liver. EASL
Clinical Practice Guidelines: Wilson’s disease. J Hepatol.
2012;56:671-85.
17. Rahimi RS, Singal AG, Cuthbert JA, Rockey DC.
Lactulose vs polyethylene glycol 3350–electrolyte solution for treatment
of overt hepatic encephalopathy: The HELP randomized clinical trial.
JAMA Intern Med. 2014;174:1727-33.
18. Als-Nielsen B, Gluud LL, Gluud C. Non-absorbable
disaccharides for hepatic encephalopathy: Systematic review of
randomised trials. BMJ. 2004;328:1046.
19. Leise MD, Poterucha JJ, Kamath PS, Kim WR.
Management of hepatic encephalopathy in the hospital. Mayo Clin Proc.
2014;89:241-53.
20. Alba L, Hay JE, Angulo P, Lee WM. Lactulose
therapy in acute liver failure. J Hepatol. 2002;36:33A.
21. Sushma S, Dasarathy S, Tandon RK, Jain S, Gupta
S, Bhist MS. Sodium benzoate in the treatment of acute hepatic
encephalopathy: A double-blind randomized trial. Hepatology.
1992;16:138-44.
22. Batshaw ML, Brusilow S, Waber L, Blom W, Brubakk
AM, Burton BK, et al. Treatment of inborn errors of urea
synthesis: activation of alternative pathways of waste nitrogen
synthesis and excretion. N Engl J Med. 1982;306:1387-92.
23. Schaefer B, Schaefer F, Engelmann G, Meyburg J,
Heckert KH, Zorn M, et al. Comparison of Molecular Adsorbents
Recirculating System (MARS) dialysis with combined plasma exchange and
haemodialysis in children with acute liver failure. Nephrol Dial
Transplant. 2011;26:3633-9.
24. Liu JP, Gluud LL, Als-Nielsen B, Gluud C.
Artificial and bioartificial support systems for liver failure. Cochrane
Database Syst Rev. 2004;1:CD003628.
25. Bourgoin P, Merouani A, Phan V, Litalien C,
Lallier M, Alvarez F, et al. Molecular absorbent recirculating
system therapy (MARS) in pediatric acute liver failure: A single center
experience. Pediatr Nephrol. 2014;29:901-8.
26. Lai YC, Huang HP, Tsai IJ, Tsau YK. High-volume
continuous venovenous hemofiltration as an effective therapy for acute
management of inborn errors of meta-bolism in young children. Blood
Purif. 2007;25:303-8.
27. Westrope C, Morris K, Burford D, Morrison G.
Continuous hemofiltration in the control of neonatal hyperammonemia: A
10-year experience. Pediatr Nephrol. 2010;25:1725-30.
28. Mazariegos G, Shneider B, Burton B, Fox IJ,
Hadzic N, Kishnani P, et al. Liver transplantation for pediatric
metabolic diseases. Mol Genet Metab. 2014;111:418-27.
29. Arnon R, Kerkar N, Davis MK, Anand R, Yin W,
González-Peralta RP, et al. Liver transplantation in children
with metabolic diseases: The studies of pediatric liver transplantation
experience. Pediatr Transplant. 2010;14:796-805
30. Stevenson T, Millan MT, Wayman K, Berquist WE,
Sarwal M, Johnston EE, et al. Long-term outcome following
pediatric liver transplantation for metabolic disorders. Pediatr
Transplant. 2010;14:268-75.
31. Dhawan A, Taylor RM, Cheeseman P, De Silva P,
Katsiyiannakis L, Mieli-Vergani G. Wilson’s disease in children: 37-year
experience and revised King’s score for liver transplantation. Liver
Transplant. 2005;11:441-8.
32. Devarbhavi H, Singh R, Adarsh CK, Sheth K, Kiran
R, Patil M. Factors that predict mortality in children with Wilson
disease associated acute liver failure and comparison of Wilson disease
specific prognostic indices. J Gastroenterol Hepatol. 2014;29:380-6.
33. Fischer RT, Soltys KA, Squires RH, Jaffe R, Mazariegos
GV, Shneider BL. Prognostic scoring indices in Wilson disease: A case
series and cautionary tale. J Pediatr Gastroenterol Nutr. 2011;52:466-9.
34. Hughes RD, Mitry RR, Dhawan A. Current status of
hepatocyte transplantation. Transplantation. 2012;93: 342-7.
35. Sokal EM. Treating inborn errors of liver
metabolism with stem cells: Current clinical development. J Inherit
Metab Dis. 2014;37:535-9.
36. Cantz T, Sharma AD, Ott M. Concise review:
Cell therapies for hereditary metabolic liver diseases - concepts,
clinical results and future developments. Stem Cells. 2015;33:1055-62.
37. De-Laet C, Dionisi-Vici C, Leonard JV, McKiernan
P, Mitchell G, Monti L, et al. Recommendations for the management
of tyrosinemia type 1. Orphanet J Rare Dis. 2013; 8:8.
|
|
|
 |
|
