|
|
Short Communications Indian Pediatrics 2008; 45:-685-688 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Langerhans’ Cell Histiocytosis: Experience from a Single Center |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Deepak Bansal, R K Marwaha, Amita Trehan, Vishal Gupta, and *Neelam Varma From the Pediatric Hematology-Oncology Unit, Department of Pediatrics,Advanced Pediatric Center and *Department of Hematology,Postgraduate Institute of Medical Education and Research, Chandigarh 160 012, India. Correspondence to: Dr R K Marwaha, Professor of
Pediatric Hematology-Oncology, Department of Pediatrics, Advanced
Pediatric Center, Postgraduate Institute of Medical Education and
Research, Chandigarh 160 012, India. Manuscript received: April 4, 2006; Initial review
completed: July 21, 2006;
Abstract
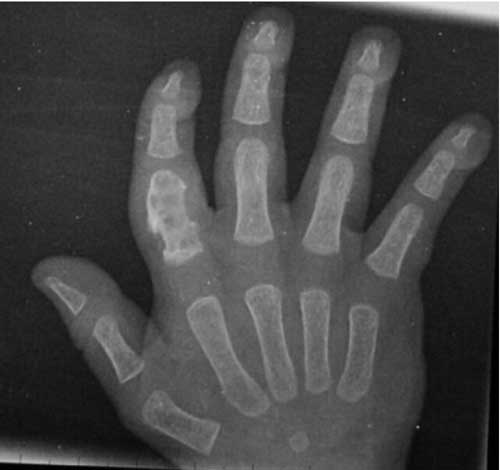
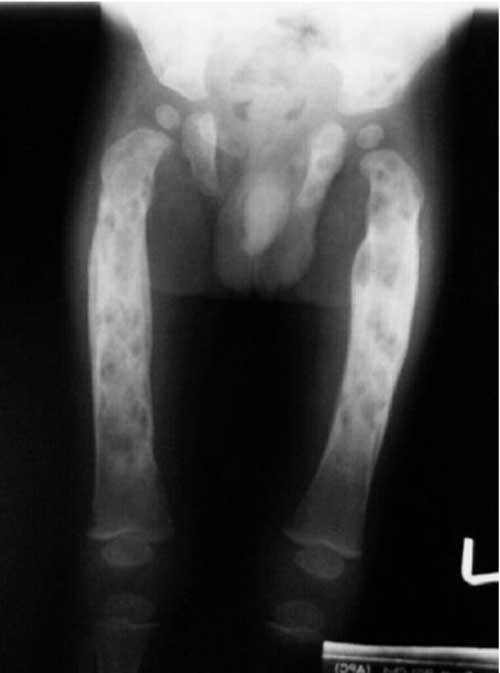
Introduction Langerhans’ cell histiocytosis (LCH) remains a versatile mimicker; diagnosis is often difficult and delayed(1). The course of the disease varies, from spontaneous resolution to a progressive multisystem disorder with organ dysfunction and potential life-threatening complications(1-4). In this communi-cation, we share our 18-year experience of managing children with LCH. Methods The case records of patients with LCH, in the period between January 1986 and December 2004, were retrieved. Confidence level for diagnosis was presumptive in majority. Supplemental stains for S-100 protein/CD1a antigen were performed 1998 onwards, for a designated/definitive diagnosis. Electron-microscopy examination for presence of Birbeck granules was not available. The extent of the disease was documented as per Gadner, et al.(5), as localized and disseminated disease. Localized disease includes single-system, single-site disease (Group 1). Patients with disseminated disease were grouped into three groups: Group A, patients with multifocal bone disease; Group B, patients with multisystem disease without organ dysfunction; and Group C, patients with multisystem disease with organ dysfunction. Organ dysfunction was defined by Lahey’s criteria(6). Children with localized disease (Groups 1 and A) were treated with prednisolone, initially at 2 mg/Kg/day for 3-4 weeks. Once disease activity subsided, prednisolone was changed to alternate day regimen. It was tapered gradually and stopped after 6-12 months. Children with disseminated disease received weekly vinblastine (6 mg/m2, for 6-8 weeks), or 3-weekly etoposide (150 mg/m2/day for 3 days, for 6 courses) along with prednisolone. Inadequate response prompted a switchover from vinblastine to etoposide and vice-a-versa. Results The age ranged between 2 months to 12 years, with a mean of 3.5±3.0 years. The M:F ratio was 6.7:1. Unisystem-unifocal disease (Group I) and multifocal bone disease (Group A) was seen in 11 (15.9%) and 10 (14.6%) cases, respectively. Disseminated disease without organ dysfunction (Group B) was seen in 11 (15.9%), whilst multisystem involvement with evidence of organ dysfunction (Group C) was documented in 37 (53.6%) children. The signs/symptoms are outlined in Table I. Distribution of bone lesions is illustrated in Table II and Fig. 1 and 2. Derangement in liver function tests was observed in 25 (36.2%) patients. Isolated elevation of alkaline phosphatase, without transaminitis was observed in 19 cases. Liver biopsy was performed in 7 children. Varying degree of fibrosis was recognized in five; two had evidence of sclerosing cholangitis. Infiltration by histiocytes was observed in 11 (23.4%) of 47 children in whom a bone marrow examination was performed. Extensive fibrosis in the marrow was recorded in one case. TABLE I Symptoms/signs of Langerhans Cell Histio-cytosis
TABLE II Distribution of Bone Lesions in Langerhans Cell Histiocytosis (n=45)
Interstitial infiltrates and reticulonodular shadows on chest X-ray were evident in 16 patients. Cystic lesions were seen in 3 and bronchiectasis and pleural effusions in one each. Vertebral collapse with surrounding granulation tissue and cord compression was observed in two cases. They presented with a gibbous deformity and neurodeficits. The mean follow-up duration was 21±28 months (range: 0-9½ years). 19 (27.5%) had a follow-up exceeding 2 years.
Groups 1 and A disease (n=21): 16 (76%) children had a complete response, without recurrences. Four (19%) patients are still on therapy, with no evidence of disease activity. A solitary case progressed to group C disease and subsequently defaulted. Two developed diabetes insipidus (DI). Group B disease (n=11): Disease free survival without recurrences was observed in 5 (45.4%), recurrence occurred in 3 cases, who subsequently defaulted, and remaining 3 patients died. Group C disease (n=37): Four families declined therapy because of financial constraints. Two defaulted after a partial response. Six cases attained a complete response without recurrences. There are 4 cases on therapy with a favorable response and a child who had ‘stable-disease’ following recurrence. There were 20 (54%) deaths, attributed to progressive disease (16), febrile neutropenia (1) and gastrointestinal bleed due to portal hypertension (3). Deaths caused by progressive disease occurred at mean duration of 3.3±3.5 months. In contrast, children with chronic liver disease and portal hypertension died 2, 5 and 7 years, following the diagnosis of LCH. DI was clinically manifest in 8 at presentation, whilst it developed in 4 children after 20±3.5 months. Lytic lesions in skull were evident in 10 children with diabetes, MRI revealed absence of posterior pituitary bright signal and stalk thickening in one child with diabetes. Two children, both initially thought to have Pott’s spine turned out to have LCH, based on CT-guided fine needle aspiration cytology. The child with involvement of the thoracic vertebrae improved completely, but the one with cervical vertebrae involvement required neurosurgical intervention. Discussion This study is a retrospective analysis of 69 children with LCH, managed in a tertiary care Institute in North India. Confidence levels for diagnosis of LCH are defined as presumptive, designated, and definitive(7). Presumptive diagnosis is based on light morphologic characteristics. Designated diagnosis is based on light morphologic features, plus two or more supplemental stains. Definitive diagnosis requires electron microscopy for demonstration of Birbeck granules and/or staining for CD1a antigen. Age distribution of cases in the series followed the typical distribution described in the literature(1,8,9); the majority of cases (79%) were below five years. The sex ratio of 6.7:1 in the series is perplexing. Earlier workers have described a slight male predominance of 1.6-2.0:1(10,11). Disseminated forms of LCH are more common in children younger than 2 years(1,12). In this study, 21 (70%) children < 2 years had Group C disease as compared to 16 (41%) children older than 2 years (P=0.22). 7 (58.3%) children > 7 years had stage I disease compared to 4 (7%) children below the age of 7 (P=0.003) Multisystem disease (stage B/C) constituted a bulk of 48 (70%) children. This reflects the clustering of ‘difficult to treat’ cases in a tertiary care center. Clinical signs/symptoms and distribution of disease in index series has largely mimicked earlier reports(1,13,14). Unusual sites of bone involvement observed included hand, wrist, foot, scapula and sternum. LCH of spine is likely to masquerade spinal tuberculosis in the endemic areas, if tissue diagnosis is not attempted. Involvement of the skin was more common in children with disseminated disease (P=0.003). Liver involvement is typically seen as a manifestation of extensive LCH(15). 19 (76%) of the 25 children with hepatic dysfunction had isolated elevation of alkaline phosphatase, without transaminitis. This suggests that portal infiltration and fibrosis leading to cholestasis is the predominant hepatic pathology in LCH, and not hepatocyte necrosis. 3 patients developed portal hypertension and succumbed several years after diagnosis. Hepatic fibrosis and portal hypertension, despite successful chemo-therapy are uncommon, though recognized sequelae(16). Although, varied treatment modalities, relatively small numbers and a large drop out rate preclude comparison of outcome between disease groups, outcome in children with Group C disease was significantly worse than that of localized disease. Contributors: DB and VG did data retrieval. DB performed literature search, data analysis and prepared the initial draft. RKM and AT critically evaluated the paper. All participated in the clinic. NV did bone marrow reporting. RKM will act as the guarantor. Funding: Nil. Conflict of interest: None stated.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
References | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
![]()