|
|
Case Reports Indian Pediatrics 2006;43:733-735 |
||||||
|
S252W Mutation in Indian Patients of Apert Syndrome |
||||||
|
K.M. Girisha
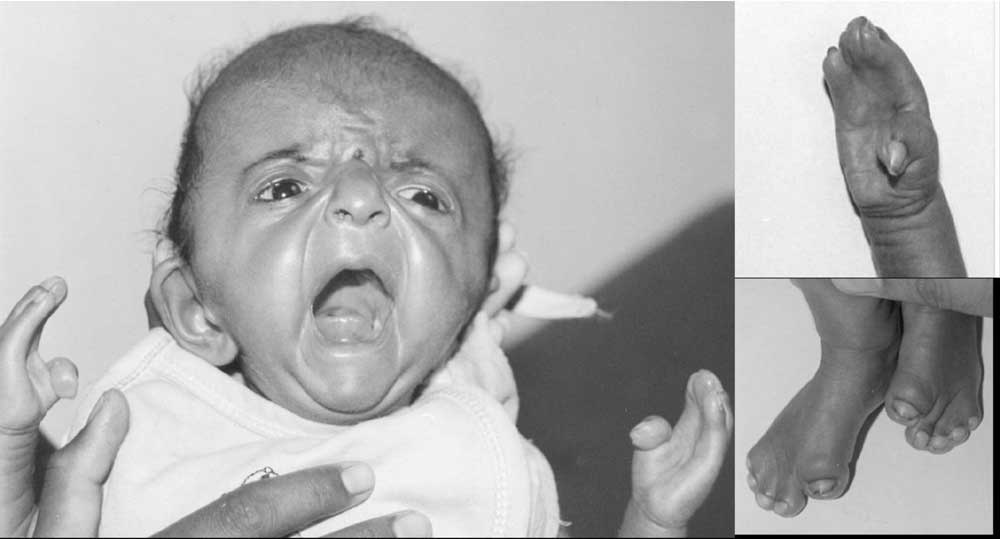
Craniosynostosis refers to premature fusion of sutures of skull. The overall birth prevalence is 343 per million newborns(1). Apert syndrome, with a birth prevalence of 15-16 per one million newborns accounts for about 4-5% of all cases of craniosynostosis. Apert syndrome is characterized by craniosynostosis, midface hypoplasia and symmetric syndactyly of hands and feet. Two common mutations in fibroblast growth factor receptor 2 (FGFR2) account for about 97% of the cases(2). The mutations S252W and P253R abolish a restriction site for MboI and BglI respectively. This provided the basis to study these mutations in two Indian patients with Apert syndrome. Subjects and Methods Case 1 Eleven months old female child was brought for evaluation of dysmorphism. She was born of a non-consanguineous marriage at full term by cesarian section in view of cephalopelvic disproportion. The child had apparently normal development. She was able to sit on her own and able to stand with support. She was babbling and understood spoken language. She did not have seizures or vomiting. Her weight was 6.5 kg (<5th percentile), length was 75 cm (50th percentile) and head circumference was 43.5 cm (within 2 SD). She had craniosynostosis, open anterior fontanel, and lambdoid suture, cleft palate, brachycephaly, bilateral proptosis, and syndactyly of both hands and feet (mitten hands). Anteroposterior diameter of skull was12.5 cm and transverse diameter was 14 cm. Deep tendon reflexes were brisk and plantar responses were extensor. Hearing and vision were apparently normal. Case 2 A one-month-old female, born to non-consanguineous couple was referred for evaluation of dysmorphism. She had age appropriate physical growth. She had bilateral proptosis, midface hypoplasia, and brachycephaly, syndactyly of hands and feet, and mild clefting of uvula. She had high arched palate and broad thumbs and great toes in addition (Fig.1).
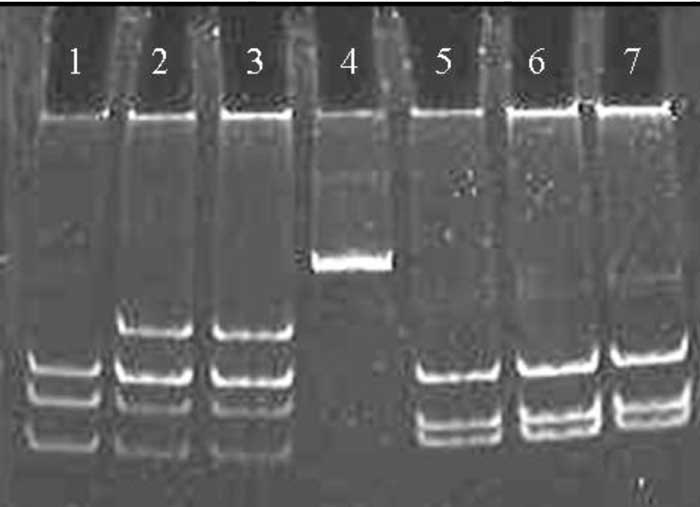
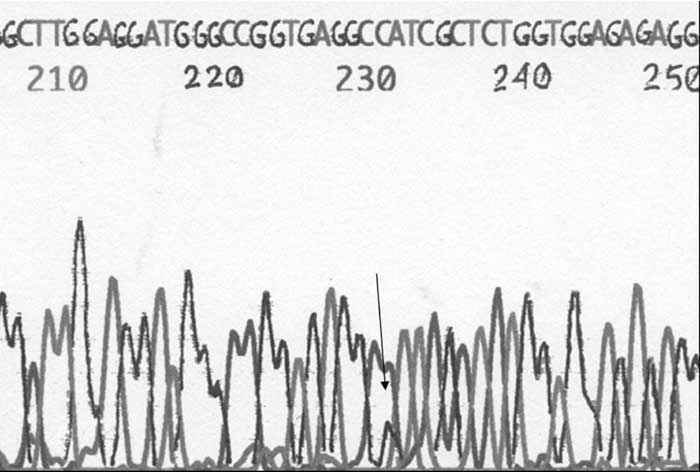
Informed consent was taken from the parents of both the subjects before performing the study. Polymerase chain reaction (PCR) followed by analysis of restriction fragment length polymorphism was done on DNA according to the published protocol(2). Both the subjects were heterozygous for S252W mutation. In both the individuals the two restriction sites were intact in the PCR product for BglI enzyme suggesting the absence of P253R mutations (Fig. 2). The mutation was confirmed by bidirectional sequencing of the PCR product using ABI 310 automated DNA fragment analyzer (Fig.3).
Discussion We analyzed mutations in two patients with Apert syndrome. As described in the literature, we also found one of the two common mutations which accounts for 71% of the cases of Apert syndrome. To the best of our knowledge, no mutations have been reported earlier in Indian literature in patients with this condition. The role of mutation detection in establishing the diagnosis is limited as the clinical phenotype is usually very characteristic. However, it is useful in situations where overlap between different craniosynostosis syndromes is noted. The same molecular methods can be used for prenatal diagnosis of the condition by chorionic villus sampling. The condition is usually due to de novo mutation and there is no significant increase in the risk of recurrence. But for many autosomal dominant and X-linked disorders germline mosaicisms are reported. The risk of recurrence due to germline mosaicism cannot be quantified, however indicates the need for prenatal diagnosis. Hence, it will be quite useful for relieving the anxiety of the couple with a previous affected child. The technique has been used earlier for prenatal diagnosis of the condition when sonographic features suggested the diagnosis of this particular condition(3). Contributors: KMG was involved in clinical evaluation of patients, carried out molecular work and drafted the manuscript. SRP conceptualized the work and was involved in clinical evaluation of patients and correction of manuscript. FK was involved in DNA sequencing and contributed to drafting of manuscript. SA guided the molecular work and DNA sequencing. She also contributed to the correction of the manuscript. She will act as guarantor. Funding: Primers and sequencing were procured from Department of Biotechnology, New Delhi funded project. Competing interests: None
| ||||||
|
References | ||||||
|
![]()