|
|
Case Report Indian Pediatrics 2004; 41:848-850 |
||||
A.S.M. Bazlul Karim
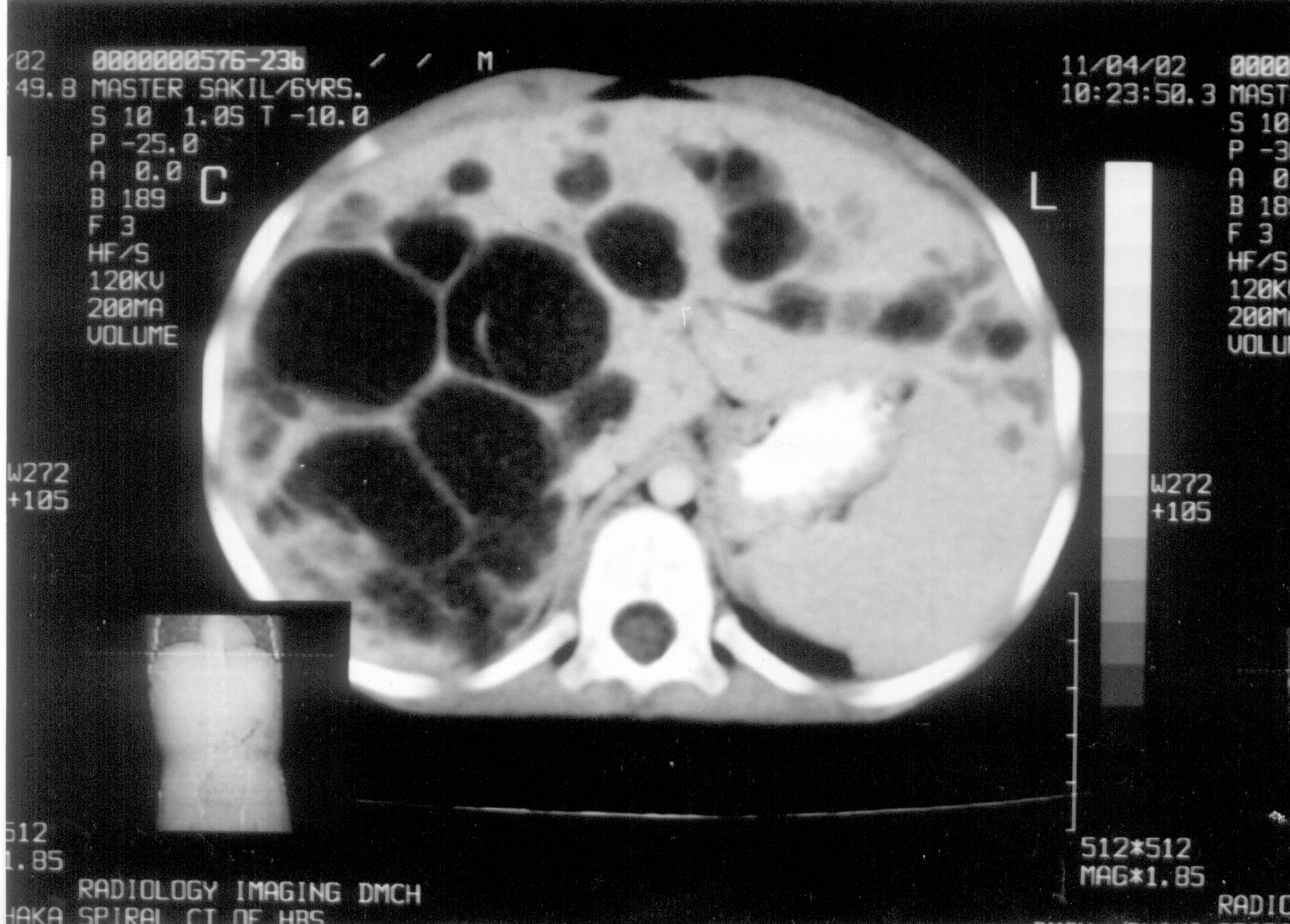
Abstract: Caroli’s disease is a rare communicating segmental or diffuse dilatation of the intrahepatic biliary tree. Cholangitis, liver cirrhosis and cholangiocarcinoma are its potential complications. A case of Caroli’s disease in a boy of 6 years with bilobal involvement presenting with intermittent abdominal pain, fever and hepatomegaly is reported here. Key words: Abdominal pain, Caroli’s disease. Cystic diseases of the bile ducts are rare congenital anomalies and are common in Japan and Asia. Most cases are diagnosed in children less than 10 years of age(1). In 1958, Jacques Caroli first described a rare congenital condition characterized by non-obstructive saccular or fusiform multi-focal segmental dilatation of the intra-hepatic bile ducts(2). The mode of inheritance is still uncertain but in majority of cases it is transmitted in an autosomal-recessive fashion(3) while in one family the mode of inheritance appeared to be autosomal dominant(4). The main clinical features are recurrent cholangitis and hepatomegaly(5). Case Report A six-year-old male boy presented with the complaints of intermittent fever and right upper quadrant abdominal pain for 3 years. Fever was associated with chills and rigor. There was no remarkable past history. He is the second son of a first degree consanguineous marriage and the other sib is healthy. On physical examination he was found non-icteric and his liver edge was 10 cm from the right costal margin in the mid-clavicular line. Liver consistency was firm and it was not tender. Spleen was not palpable. Other physical signs were unremarkable. Laboratory investigations showed poly-morphonuclear leukocytosis with moderate elevation of ESR. Liver function tests like ALT, bilirubin, PT, alkaline phosphatase and albumin were within normal limit. Routine urine examination, kidney function tests and x-ray chest were normal. Ultrasound (US) and computed tomography (CT) of abdomen (Fig. 1) showed multiple cystic dilated bile ducts with bridge formation within the liver occupying the both lobes but more in the right lobe. No feature of hepatic fibrosis was seen and both the kidneys were normal. Cholangiography though helpful, was not possible due to technical reason. Liver biopsy, for exclusion of associated congenital hepatic fibrosis (CHF), was not done because CT scan was not suggestive and there was absence of clinical evidence of portal hypertension. A diagnosis of isolated Caroli’s disease was made and the child was treated conservatively with antibiotics. Partial hepatic lobectomy was advised but parents did not agree. The parents were subsequently counseled, properly advised and asked to come for regular follow-ups.
Discussion There are two forms of Caroli’s disease, one associated with congenital hepatic fibrosis and a simpler form occurring alone. The former, called Caroli’s syndrome is associated with portal hypertension. The later, known as Caroli’s disease, may be associated with autosomal recessive polycystic kidney disease(6) or rarely with autosomal dominant polycystic kidney disease(7). Caroli’s disease has also been reported in patient with choledochal cysts for which reason some authorities classify it as a type of choledochal cyst(8). Caroli’s disease may be localized to one lobe of liver or may be diffuse. It results from an arrest in ductal plate remodeling at the level of the larger intra-hepatic bile ducts(3). Caroli’s disease usually presents with intermittent abdominal pain and hepato-megaly. Cholangitis, cholelithiasis, bilary abscess, septicemia, liver cirrhosis and cholangiocarcinoma are all its potential complications. Malignant complication (cholangiocarcinoma) occurs in approxi-mately 7% of cases(1) and is due to prolonged exposure of the ductal epithelium to high concentration of unconjugated secondary bile acids(9). The diagnosis of Caroli’s disease rests on demonstrating that the cystic lesions are in continuity with the bilary tree. It can be done by imaging studies such as abdominal USG, CT scan, isotope scan, ERCP, PTC and MRCP. These studies demonstrate irregular cystic dilatation of the large proximal intra-hepatic bile ducts with normal ducts in between. The treatment of Caroli’s disease depends on the clinical features and the location of the biliary abnormalities. Cholangitis is treated with appropriate antibiotics. In case of intrahepatic cholelithiasis litholytic therapy with urso-deoxy cholic acid (UDCA) is indicated(10). When the ductal abnormalities are localized to one lobe, lobectomy relieves symptoms and appears to remove the risk of malignancy. In case of diffuse involvements of both lobes of liver, treatment options include conservative management, endo-scopic therapy (sphincterotomy for clearance of intra-hepatic stone), internal biliary bypass procedures and in carefully selected cases liver transplantation(3). Those who can not be operated radically should have regular clinical follow ups including ultrasound and liver biopsy as necessary. Further family studies are needed in all cases to exclude the autosomal dominant mode of inheritance(4). Abdominal pain is a common pediatric problem and though it is a rare congenital anomaly one should keep in mind the possi-bility of Caroli’s disease in its differential diagnosis especially in children who present with intermittent fever and hepatomegaly along with abdominal pain. Funding: None. Competing interests: None stated.
| ||||
|
|
![]()