Jagruti P Sanghvi , Surekha B Rajadhyaksha and Meher
Ursekar*
From the Epilepsy Clinic, Department of
Pediatrics, Bai Jerbai Wadia Hospital for Children,Parel,
Mumbai-400012 and *Department of Radiology, Bombay Hospital, Marine
Lines,Mumbai, India.
Correspondence to: Dr. Jagruti P. Sanghvi,501,
Gresil Apartments, Irla Lane, Vile Parle (W),
Mumbai- 400 056. India. Email address:
dr_jagruti@hotmail.com
Manuscript received: July 28, 2003, Initial review
completed: September 9, 2003;
Revision accepted: January 20, 2004.
Abstract:
This study was conducted in a tertiary
pediatric epilepsy clinic to ascertain the spectrum of
development malformations in children, with seizures. Seventy
Six Children (0-12 yr) with seizures and CNS malformations based
on neuroimaging were included. Observed anomalies included
dysgenetic corpus callosum (DCC), lissencephaly, focal cortical
dysplasia (FCD), pachygyria,polymicrogyria, heterotopia,
schizencephaly, holoprosencephaly, hemimegalencephaly, and
phakomatoses like tuberous sclerosis, Sturge Weber syndrome and
linear cutaneous nevus syndrome. Seizure semiology varied in all
categories. Microcephaly, developmental delay and tone
abnormalities were common clinical findings. 60.5% cases
presented in infancy. The characteristic EEG features provided a
clue to the diagnosis of anomalies like lissencephaly, agenesis
of corpus callosum and alobar holoprosencephaly.
Key words: CNS malformations, EEG, Epilepsy,
Neuroimaging.
Developmental CNS malformations are a complex
group of congenital malformations often presenting with variable
neuro-developmental dysfunction and seizures(1). Computed
tomography (CT) scan and magnetic resonance imaging (MRI) have
revolutionized our understanding of these malformations, providing
a good anatomic diagnosis. However, accurate pathological
diagnosis can be done by histopathology alone. We conducted a
study to evaluate the entire spectrum of CNS malformations in
children with seizures, as diagnosed by neuroimaging.
Subjects and Methods
Over a 5 year priod from 1996 to 2001, 1330
children were enrolled from the epilepsy clinic of a tertiary care
pediatric hospital. These children were evaluated for age of
seizure onset, sex, risk factors, family history, developmental
delay, dysmorphic features, seizure semiology, neurological
examination and EEG. Based on this, patients were advised
neuroimaging. 76 patients were diagnosed as having developmental
CNS malformations by neuroimaging. Of these, CT scan could
diagnose brain malformations in 33 patients and 27 patients
underwent MRI only; 16 cases underwent both CT scan and MRI. The
same neuroradiologist reviewed all the scans. Video EEG was
performed in cases with doubtful seizure semiology, to identify
the seizure type and concomitant EEG abnormality. In each patient,
an attempt was made to correlate the epileptic seizures with the
location, extent and type of CNS malformation and EEG features.
Results
Seventy six cases were identified to have
epileptogenic brain malformations based on neuroimaging. These
malformations included dysgenetic corpus callosum (DCC) (n = 19),
lissencephaly(n = 9), focal cortical dysplasia (FCD) (n = 9),
pachygyria (n = 6), poly-microgyria (n = 3), heterotopia(n = 4),
schiz-encephaly(n = 2), holoprosencephaly(n = 4),
hemimegalencephaly(n = 1), and phako-matoses like tuberous
sclerosis (TS)(n = 15), Sturge Weber syndrome (SWS)(n = 3) and
linear cutaneous nevus syndrome (LCNS) (n = 1). There was a male
preponderance (60.5%) in our study, mainly in pachygyria and
heterotopias (100%). Patients with Aicardi syndrome were females
(3/3). CT scan could mainly diagnose phakomatoses and
holoprosencephaly (4/4) and hemi-megalencephaly (1/1). Sixteen
patients with neuronal migrational abnormalities, previously not
diagnosed by CT scan were recognized by a subsequent MRI scan.
Forty six out of 76 (60.5%) cases had their
first seizure in infancy, and included 13 neonates. Patients with
holoprosencephaly (4/4) and hemimegalencephaly (1/1) presented in
neonatal period; generalized disorders like lissencephaly and DCC
in infancy whereas focal disorders like polymicrogyria, hetero-topia
and phakomatoses like SWS had a later age of seizure onset.
The seizures, as per the classification by the
International League Against Epilepsy(2), were partial in 27/76
(35.6%) and generalized in 31/76 (40.7%) cases. 15/76 (19.7%)
presented with infantile spasms, of which 7 had TS, 5 had
lissencephaly and 3 had DCC. Two patients had unclassifiable
seizures. Multiple seizure types were seen in 38.2% cases.
Six patients were born of a consan-guineous
marriage, including 2 siblings with lissencephaly and six had a
family history of epilepsy.
At presentation, 15/76 (19.7%) patients had
normal neurodevelopmental assessment, 49/76 (64.5%) patients had
global delay in milestones, 5 had motor delay, 2 had isolated
speech delay and 4 patients (5.3%) showed neuro-regression. 22/76
(28.9%) patients exhibited microcephaly, whereas macroce-phaly was
seen with hemimegalencephaly (1/1). Dysmorphic features (n = 10)
were clue to syndromes like Miller-Dieker, Aicardi, Sotos and
Pierre-Robin and 4 patients had holoprosencephalic facies.
Neuro-cutaneous markers like ash-leaf macules, adenoma sebaceum,
shagreen patch, café-au-lait spots, and nevus were seen in 17
patients. Focal neurological deficit and tone abnormalities were
observed in 13 and 29 children respectively.
Eighty per cent of DCC and 55.5% cases with
lissencephaly had other associated CNS abnormalities, which
included colpocephaly, Dandy-Walker variants, cysts, heterotopia,
FCD, polymicrogyria and pachygyria. All patients with
holoprosencephaly and hemi-megalencephaly had associated
pachygyria.
As shown in Table I, 64/76 (84.2%) patients had
abnormal EEG, whereas 12 /76 (15.8%) had a normal EEG. EEG of
17.1% cases showed modified hypsarrhythmia, presented clinically
with infantile spasms and constituted the West syndrome. 11/19
(58%) patients with DCC showed asymmetry between the two
hemispheres on EEG (Fig. 1). Two cases with alobar
holoprosencephaly exhibited a unique type of EEG abnormality
showing frequent, asynchronous sharp waves, spike, and spike-wave
and polyspike-wave complexes over both frontal regions, with
decreasing gradient of potentials over the frontal to occipital
leads. They also showed high amplitude rhythmic delta activity in
the centro-temporal region with almost isoelectric record
posteriorly. High amplitude fast rhythms of a- and b-activity was
identified in patients with lissencephaly (Fig. 2). Both patients
with schizencephaly showed asymmetry between the 2 hemispheres
with epileptiform discharges in the hemisphere with the cleft and
absence of secondary generalization.
 |
 |
|
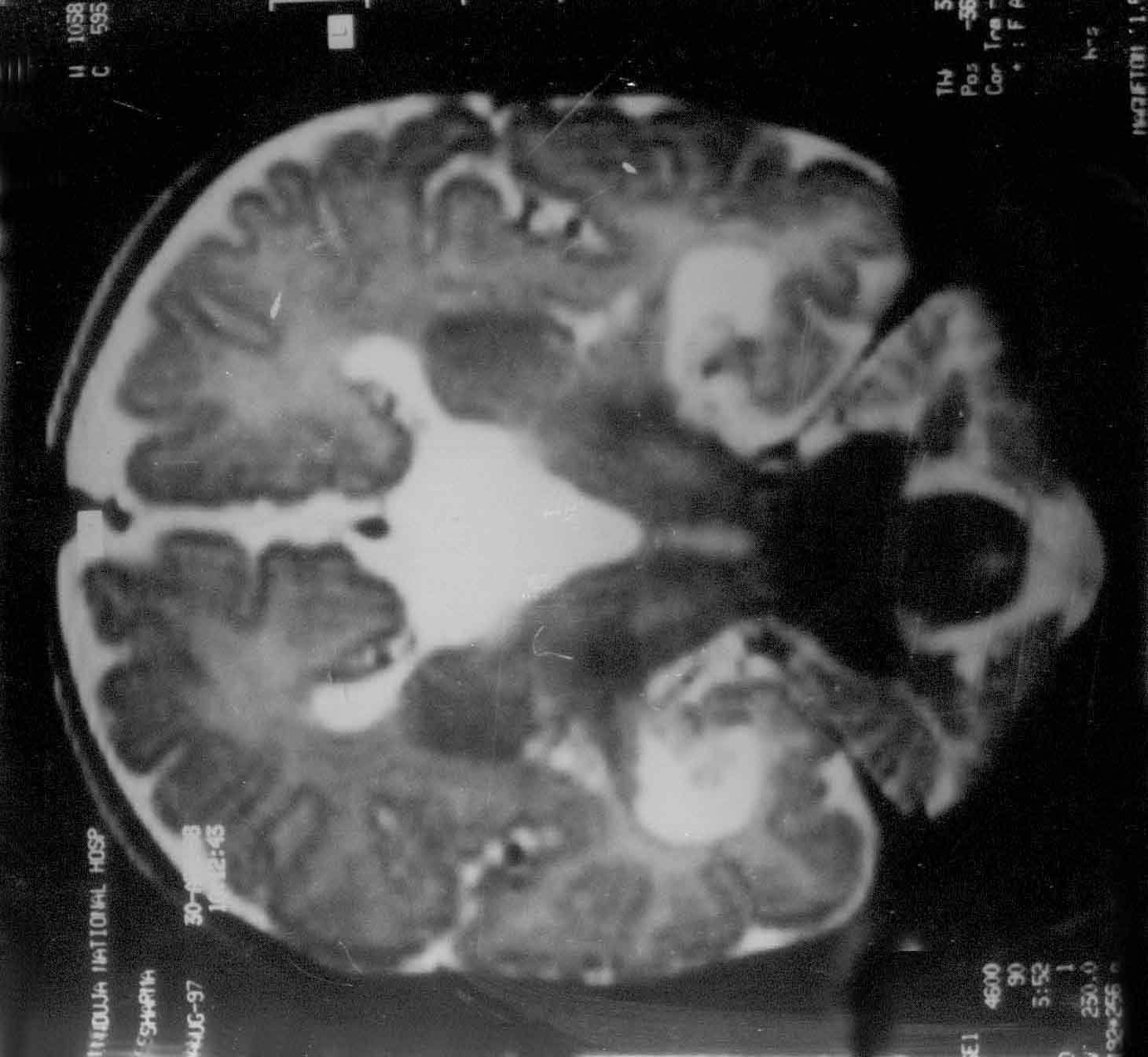
Fig. 1. Agenesis of
corpus callosum. T2W coronal MR image revealing a typical
"Viking-helmet" deformity of the lateral ventricle and the
dilated and elevated 3rd ventricle. Simultaneous EEG Tracing
shows asymmetry between the 2 hemispheres. |
 |
 |
|
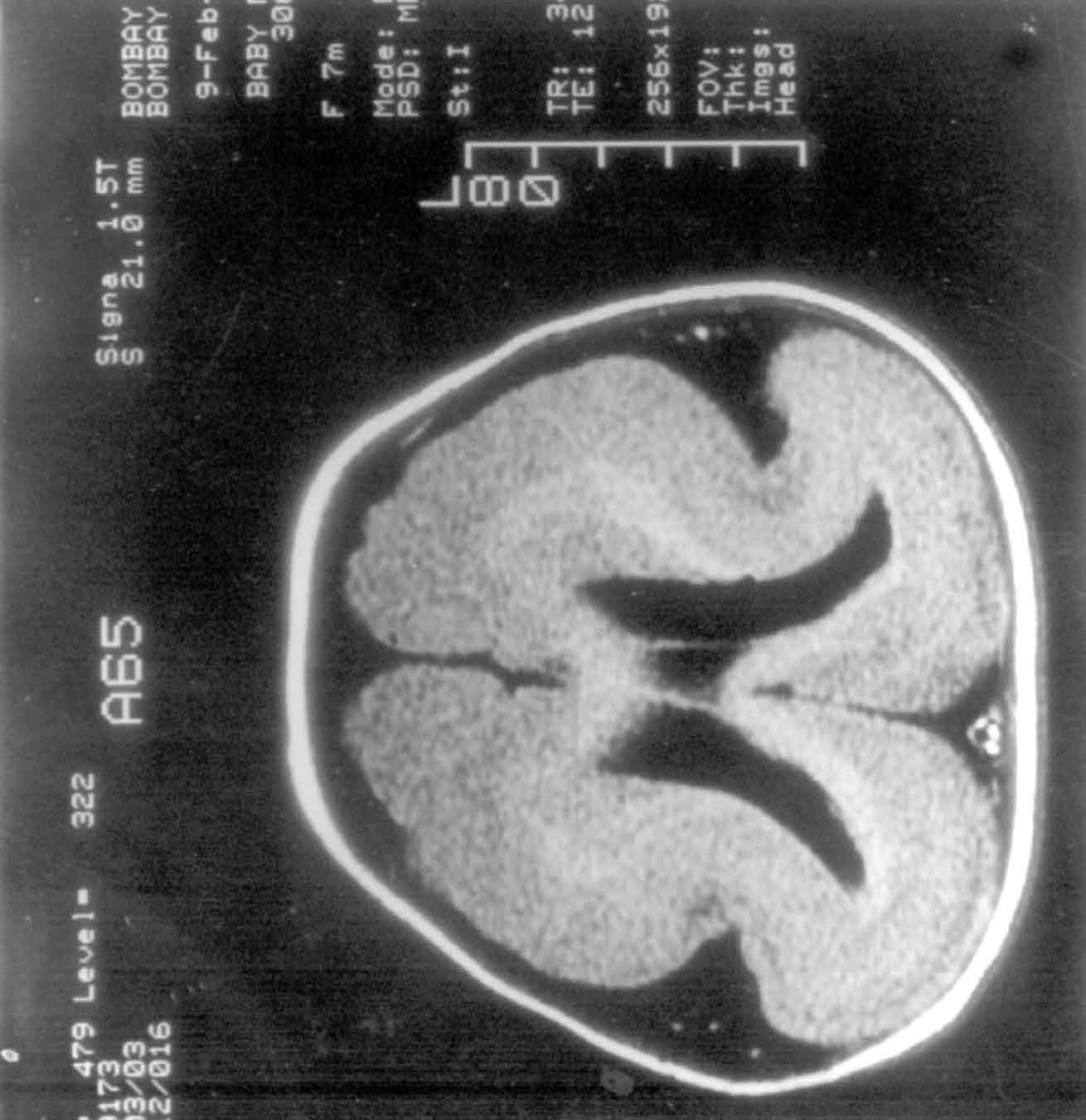
Fig. 2. Type I Lissencephaly. Axial MR
image showing the classic "figure of 8" appearance and its
EEG tracing showing high amplitude fast a and ß activity
with a mixture of high amplitude q and d rhythm. |
Table I
EEG Features of Developmental Malformations.
Category
(No. of patients) |
Generalised
neuronal
hyper-excitability |
Focal
epileptic
form
discharges |
Secondary
generali-sation |
Burst
suppre-ssion |
Asym-metry |
Hypsar-rhythmia |
Low
to absent
brain
activity |
Normal |
DCC (19)
|
1
|
7
|
2
|
1
|
11
|
3
|
-
|
1
|
Lissencephaly (9)
|
3
|
3
|
-
|
-
|
2
|
3
|
-
|
-
|
Pachygyria (6)
|
-
|
4
|
-
|
-
|
-
|
-
|
-
|
2
|
Polymicrogyria (3)
|
1
|
2
|
-
|
-
|
-
|
-
|
-
|
-
|
Heterotopia (4)
|
-
|
3
|
-
|
-
|
1
|
-
|
-
|
-
|
FCD (9)
|
-
|
1
|
3
|
1
|
1
|
1
|
-
|
3
|
Schizencephaly (2)
|
-
|
1
|
-
|
-
|
2
|
-
|
-
|
-
|
TS(15)
|
2
|
-
|
2
|
-
|
-
|
6
|
-
|
5
|
SWS (3)
|
-
|
1
|
-
|
-
|
1
|
-
|
-
|
1
|
LCNS (1)
|
-
|
-
|
-
|
-
|
1
|
-
|
-
|
-
|
Holoprosencephaly (4)
|
2
|
-
|
-
|
-
|
-
|
-
|
2
|
-
|
Hemimegalencephaly (1)
|
-
|
-
|
-
|
1
|
-
|
-
|
-
|
-
|
* Some patients had more than one EEG abnormality.
Discussion
Our study illustrates the spectrum of CNS
malformations in 76 pediatric epileptic patients, diagnosed by
neuroimaging, with relevant clinical features and EEG findings.
Unfortunately, there are not many comparable studies below 12-yr
age group encompassing the entire spectrum of brain malformations.
Kapoor, et al.(3), reported developmental lesions only in the 0-3
year age group, these included atrophy, dilated ventricles,
aqueductal stenosis and porencephaly, besides other structural
lesions. Raymond, et al.(4) studied the spectrum of cortical
anomalies in the adult population. Many of our references are
therefore, limited to individual studies on subcategories of these
malformations.
CT scan can easily identify phakomatoses due to
its calcified lesions and gross anomalies like holoprosencephaly
and hemimegalen-cephaly. However, neuronal migrational anomalies
are better identified by MRI due to its multiplanar imaging and
higher anatomic resolution (5).
Abnormalities of gyration and DCC were amongst
the commonest anomalies in our study, similar to the one on adult
population by Raymond, et al.(4). However, they had large number
of heterotopias (28%) as compared to 5.3% in our study, suggesting
that patients with heterotopias may present in adulthood. TS
formed 19.7% of our cases in comparison with 5% in the same adult
study.
60.5% of our patients presented in infancy,
including 43.4% in the 1 month-1 year age group, which is in
concordance with the Indian study by Kapoor, et al.(3). All 4
patients with heterotopia (3 with peri-ventricular and 1 with
subcortical heterotopia) were male, which is in contrast to
previous studies(4,6,7), where a female preponderance was
definitely noted in periventricular heterotopias suggesting an
X-linked dominant inheritance. However, no such sex preponderance
is noted for subcortical hetero-topias. Probably, the male
preponderance in our study reflects the referral pattern in our
country.
The seizure semiology in our series varied
widely and did not always appear to predict either the location or
morphology of the cortical malformation, which is in agreement
with other authors(8,9). The same CNS malformation was associated
with both partial and generalized seizures. However, focality of
the malformation was an important factor in the causation of
partial seizures in cases of FCD, pachygyria, SWS and LCNS. 19.7%
seizures were described as infantile spasms, an age-related
seizure pattern, and are more common in a pediatric study. 8/15
patients with TS had infantile spasms, which is the commonest
seizure type described in TS(10).
64.5% of our patients had global delay compared
to 10% in the adult study by Raymond, et al.(4), reflecting that
earlier onset of seizures denote severity of malformation. All
cases of lissencephaly showed global developmental delay, which is
comparable to the study by Barkovich, et al.(11). 5.3% patients
had neuroregression, which could be attributed to recurrent,
refractory seizures. Microcephaly is a well-documented finding in
congenital CNS malformations. Macrocephaly was observed in our
patient with hemimegalencephaly, consistent with Kalifa, et
al.(12).
EEG is an important tool in the study of
childhood epilepsy and can provide a clue to the diagnosis of some
developmental CNS malformations. Burst suppression pattern and
hypsarrhythmias are age-related EEG patterns that present in
infancy and may get modified later on. 58% patients with DCC
showed characteristic interhemispheric asynchrony similar to
previous studies(13-15). The asynchronous EEG is later replaced by
multifocal epileptiform abnormalities as seen in 15.8% of our
patients. Isoelectric or almost flat EEG changes may be described
in lesions like hydranencephaly, porencephaly, hema-tomas,
hygromas, etc. However, a combina-tion of inactivity posteriorly
and bizarre activity anteriorly is seen exceptionally in alobar
holoprosencephaly(14). High ampli-tude fast a- and b-rhythms
alternating with q and d-rhythms are classical of
liss-encephaly(4). Granata, et al.(16) explained that the absence
of generalization in EEG of patients with schizencephaly might be
due to the cleft-induced anatomical rearrangements of cortico-cortical
and cortico-subcortical pathways linking the 2 hemispheres thus
preventing bilateral diffusion of the epileptiform discharges.
However, absence of generalization per se can also be seen in
other conditions like FCD, focal atrophy, etc.
Contributors: JPS collected, analyzed and
inter-preted the data and drafted the article. SBR conceived and
designed the study, supervised the data collection, and
participated in analysis and interpretation of data. MU reported
all the CT and MRI scans. SBR revised the manuscript critically
and will act as a guarantor for the same.
Funding: None.
Competing interests: None stated.
|
Key
Messages |
• Developmental CNS malformations must be suspected in a child
who presents with seizures, neurodevelopmental delay and/or
dysmorphic features and neuro-cutaneous markers.
• Although EEG can provide a clue to the diagnosis of these
anomalies, neuroimaging is required for accurate anatomic
diagnosis.
|
|
|
1. Osborn AG. Normal brain development
and general classification of congenital malformations.
In: Osborn AG, editor. Diagnostic Neuroradiology. 1st
ed. St Louis, Mosby-Yearbook, 1994. p 3-14.
2. International League Against Epilepsy.
Proposal for revised classification of epilepsies and
epileptic syndromes. Epilepsia 1989; 30; 389-399.
3. Kapoor M, Talukdar B, Chowdhury V,
Puri V, Rath B. Intracranial structural lesions in young
epileptics: a computed tomographic study. Indian Pediatr
1998; 35: 537-541.
4. Raymond AA, Fish DR, Sisodiya SM,
Alsanjari N, Stevens JM, Shorvon SD. Abnormalities of
gyration, heterotopias, tuberous sclerosis, focal cortical
dysplasia, microdysgenesis, dysembryoplastic neuro-epithelial
tumor and dysgenesis of the archicortex in epilepsy:
Clinical, EEG and neuroimaging features in 100 adult
patients. Brain 1995; 118: 629-660.
5. Kuzniecky RI. MRI in developmental
disorders of the cerebral cortex. Epilepsia 1994; 35:
S44-S56.
6. Dubeau F, Tampieri D, Lee N, Andermann
E, Carpenter S, Leblanc R et al. Periventricular and
subcortical nodular heterotopia. A study of 33 patients.
Brain 1995; 118: 1273-1281.
7. Huttenlocher PR, Taravath S, Mojtahedi
S. Periventricular heterotopia and epilepsy. Neurology 1994;
44: 51-55.
8. Palmini A, Andermann F, Olivier A,
Tampieri D, Robitaille Y. Focal neuronal migrational
disorders and intractable partial epilepsy: a study of 30
patients (review). Ann Neurol 1991; 30: 741-749.
9. Chugani H, Shields WD, Shewmon DA,
Olson DM, Phelps ME, Peacock WJ. Infantile spasms: I. PET
identifies focal cortical dysgenesis in cryptogenic cases
for surgical treatment. (see comments). Ann Neurol 1990; 27:
406-413.
10. Berg BO. Neurocutaneous syndrome:
Phakomatoses and allied conditions. In: Swaiman KF,
Ashwal S, editors. Pediatric Neurology-Principles and
Practice. 3rd ed. St. Louis Mosby Inc, 1999. p. 530-550.
11. Barkovich AJ, Koch TK, Carrol CL. The
spectrum of lissencephaly: report of 10 patients analyzed by
MRI. Ann Neurol 1991; 30: 139-146.
12. Kalifa GL, Chiron C, Sellier N.
Hemimegalencephaly: MRI in 5 children. Radiology 1987; 165:
29-33.
13. Duchowny M, Harvey AS. Pediatric
epilepsy syndromes: an update and clinical review. Epilepsia
1996; 37: S26-36.
14. Shah KN, Rajadhyaksha SB, Shah VS,
Wakde M. EEG recognition of holoprosencephaly and Aicardi
syndrome. Indian J Pediatr 1992; 59: 103-108.
15. Fariello RG, Chun R, Doro JM. EEG
recognition of Aicardi syndrome. Arch Neurol 1977; 34:
563-566.
16. Granata T, Battaglia G, D’Incerti L, Francesschetti
S, Spreafic R, Battino D, et al. Schizencephaly:
Neuroradiologic and epileptogenic findings. Epilepsia 1996;
37: 1185-1193.
.
|
