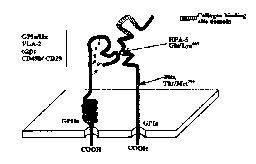
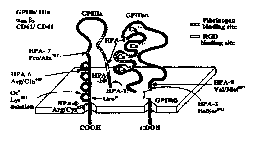
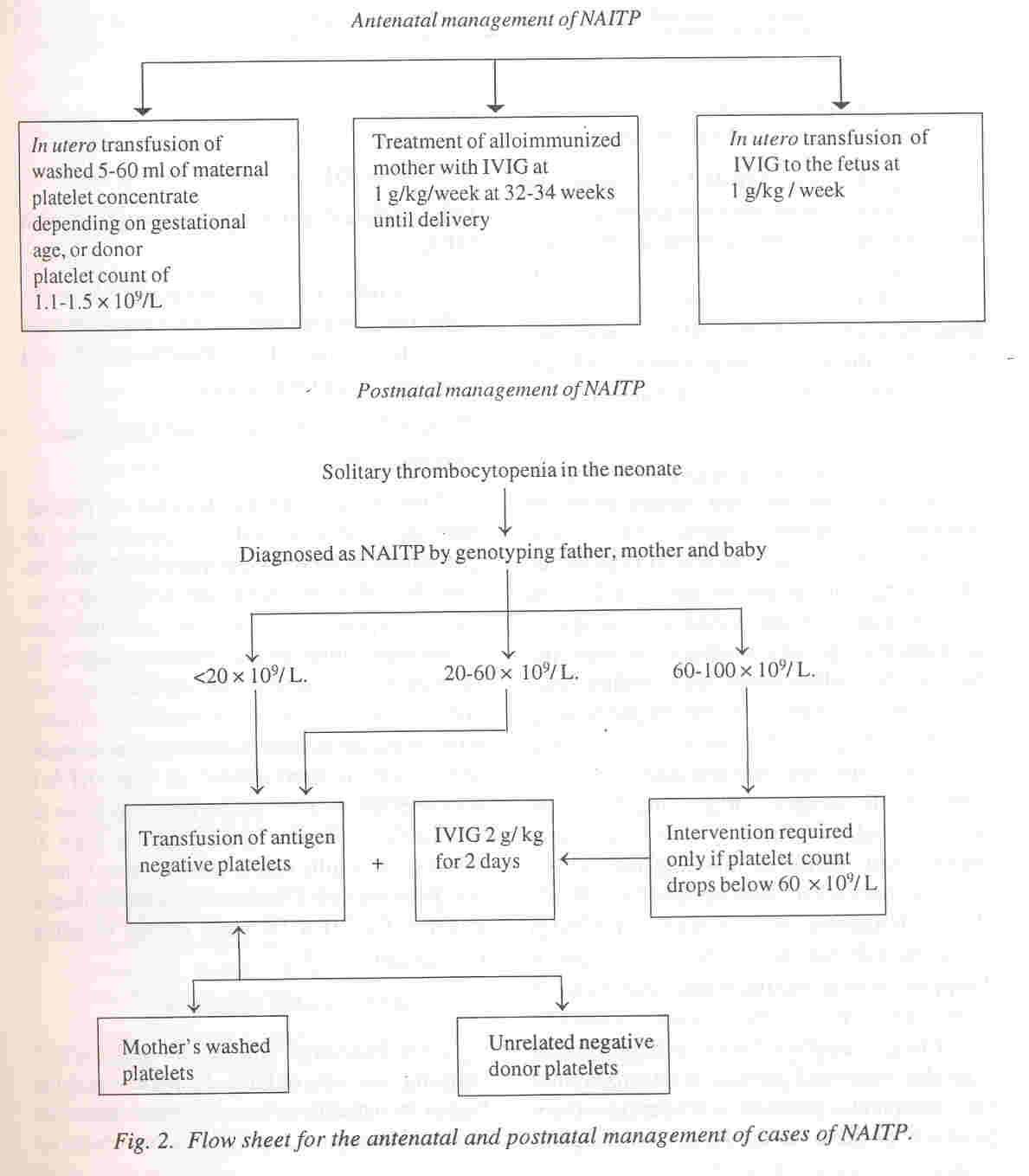
1. Newman PJ, Valentin N. Human platelet alloantigens: recent
findings, new pers-pectives. Thromb Hemost 1995 ;74: 234-239.
2. Van Loghem JJ, Dorfmeijer H, Vander Hart
M, Schreuder F. Serological and genetical studies on a
platelet antigen (Zw). Vox Sang 1959; 4: 161-169.
3. Kulkarni B, Mohanty D, Ghosh K, Pawar A,
Khare A. Frequency distribution of antigens in the human
platelet antigen-1 system in western Indian population.
Transfusion 2002; 42: 317-320.
4. Blanchette VS, Johnson J, Rand M. The
management of alloimmune neonatal thrombo-cytopenia. Bailliers
Clin Hematol 2000; 13: 365-390.
5. von dem Borne A, Decary F. ICSH/ ISBT
working party on platelet serology. Vox Sang 1990; 58: 176.
6. Smith OP. Inherited and congenital
thrombo-cytopenia. In: Pediatric Hematology, 2nd edn.
Eds. Lilleyman JS, Hann IM, Blanchette VS. London: Churchill
Livingstone 1999, p 419-435.
7. Kaplan C, Morel-Kopp MC, Kroll H, Kiefel
V, Schlegel N, Chesnel N, et al. HPA-5b (Bra) neonatal
alloimmune thrombocytopenia. clinical and immunological
analysis of 39 cases. Br J Haematol. 1991; 78: 425-429.
8. Matsui K, Ohsaki E, Goto A, Koresawa M,
Kigasawa H, Shibata Y. Perinatal intra- cranial hemorrhage due
to severe neonatal alloimmune thrombocytopenic purpura (NAITP)
associated with anti-Yukb (HPA-4a) antibodies. Brain Dev 1995;
17: 352-355.
9. Mueller-Eckhardt C, Kiefel V, Grubert A,
Weisheit M, Mueller- Eckhardt G, Kiefel V, et al. 348
cases of suspected neonatal alloimmune thrombocytopenia.
Lancet 1989; 1: 363-366.
10. Mueller-Eckhardt C, Kiefel V. HLA-DRw6,
a new immune response marker for immunization against the
platelet alloantigen Bra . Vox Sang 1986; 50: 94-99.
11. Jallu V, Meunier M, Brement M, Kaplan
C. A new platelet polymorphism Duva+, localized within the RGD
binding domain of glycoprotein IIIa, is associated with
neonatal thrombo-cytopenia. Blood 2002; 99: 4449-4456.
12. Dreyfus M, Kaplan C, Verdy E, Schlegel
N, Zaleski ID, Tchernia G and the Immune Thrombocytopenia
Working Group. Frequency of immune thrombocytopenia in new-borns:
a prospective study. Blood 1997; 89: 4402- 4406.
13. Bussel J, Kaplan C. The fetal and
neonatal consequences of maternal alloimmune thrombocytopenia.
Bailliers Clin Hematol 1998; 11: 391-408.
14. Murphy MF, Waters AH, Doughty HA,
Hambley H, Mibashan RS, Nicolaides K, et al. Antenatal
management of fetomaternal alloimmune thrombocytopenia- report
of 15 affected pregnancies. Trans Med 1994; 4: 281-292.
15. Kunicki TJ, Beardsley DS. The
alloimmune thrombocytopenias: neonatal alloimmune
thrombocytopenic purpura and post-trans-fusion purpura. Prog
Hemostas Thromb 1989; 9: 203-232.
16. Murphy MF, Hambley H, Nicolaides K,
Waters AH. Severe fetomaternal alloimmune thrombocytopenia
presenting with fetal hydrocephalus. Prenat Diagn 1996; 16:
1152-1155.
17. Chalmers EA, Gibson BES. Hemostatic
problems in the neonate. In: Pediatric Hematology, 2nd
edn. Lilleyman JS, Hann IM, Blanchette VS. Eds. London:
Churchill Livingstone 1999; p 651-678.
18. Davidson JE, McWilliam RC, Evans TJ,
Stephenson JB. Porencephaly and optic hypoplasia in neonatal
isoimmune thrombo-cytopenia. Arch Dis Child 1989; 64: 858-860.
19. Muller-Eckhardt C, Kayser W, Forster C,
Muller Eckhardt G, Ringenberg C. Improved assay for detection
of platelet-specific PLA1 antibodies in neonatal immune
thrombo-cytopenia. Vox Sang 1982; 43: 76-81.
20. Kaplan C, Daffos F, Forestier F.
Management of alloimmune thrombocytopenia: antenatal diagnosis
and in utero transfusion of maternal platelets. Blood
1988; 72: 340-343.
21. Mcfarland JG, Frenzke M, Aster RH.
Testing of maternal sera in pregnancies at risk for neonatal
alloimmune thrombocytopenia. Transfusion 1989; 29: 128-133.
22. Elgelfriet CP, Reesink HW, Kroll H,
Giers G, Bald R, Kanhai H, et.al. Prenatal management
of alloimmune thrombocytopenia of the fetus. Vox Sang 2003;
84: 142-149.
23. Spencer JA, Burrows RF. Feto-maternal
alloimmune thrombocytopenia: a literature review and
statistical analysis. Aust NZ Obstet Gynaecol 2001; 41: 45-55.
24. Radder CM, Brand A, Kanhai HHH. Will it
ever be possible to balance the risk of intracranial
hemorrhage in fetal or neonatal alloimmune thrombocytopenia
against risk of treatment strategies to prevent it? Vox Sang
2003; 84: 318-325.
25. Williamson LM, Hackett G, Rennie J,
Palmer CR, Maciver C, Hadfield R, et al. The natural
history of fetomaternal alloimmunization to the
platelet-specific antigen HPA-1a (PLA1, Zwa) as determined by
antenatal screening. Blood 1998; 92: 2280-2287.
26. Vain NE, Bedros AA. Treatment of
isoimmune thrombocytopenia of the newborn with transfusion of
maternal platelets. Pediatrics 1979; 63: 107-109.
27. Baumann MA, Menitore JE, Aster RH,
Anderson T. Urgent treatment of idiopathic thrombocytopenic
purpura with single-dose gamma globulin infusion followed by
platelet transfusion. Ann Int Med 1986; 104: 808-809.
28. Shulman NR, Aster RH, Leiitner A,
Hiller MC. Immunoreaction involving platelets. V.
Post-transfusion purpura due to a complement fixing antibody
against a genetically controlled platelet antigen: A proposed
mechanism for thrombocytopenia and its relevance in
"autoimmunity". J Clin Invest 1961; 40: 1597-1620.
29. Mueller-Eckhardt C. HLA-B8 antigen and
anti-PLA1 alloimmunization. Tissue Antigens 1982; 19: 154-158.
30. Tazzari PL, Ricci F, Tassi C, Bontadini
A, Fruet F, Conte R. Alloimmunization against human platelet
antigen 2 (HPA-2) in a series of multi-transfused beta-thalassemia
patients. Hematologica 1998; 83: 765- 766.
31. Taaning E, Skov F. Elution of anti- Zwa
(PLA1) from autologous platelets after normalization of
platelet count in post-transfusion purpura. Vox Sang 1991; 60:
40-44.
32. Pober M, Kyrle AP, Panzer S. Genotyping
provides a reliable tool for the determination of the platelet
antigen system HPA-1 in Glanzmann’s thrombasthenia. Br J
Hematol 1993; 85: 112-115.
33. Kunicki JT, Aster RH. Deletion of the
platelet- specific alloantigen PLA1 from platelets in
Glanzmann thrombasthenia. J Clin Invest 1978; 61: 1225-1231.
34. Croft SA, Samani NJ, Teare MD, Hampton
KK, Steeds RP, Channer KS, et al. Novel platelet
membrane glycoprotein VI dimorphism is a risk factor for
myocardial infarction. Circulation 2001; 104: 1459-1463.
35. Ghosh K, Kulkarni B, Nair S, Shetty S,
Mohanty D. Human platelet alloantigen polymorphism in
Glanzmann thrombasthenia and its impact on the severity of the
disease. Br J Haematol 2002; 119: 348- 353.
36. Carter AM, Ossei-Gerning N, Wilson IJ,
Grant PJ. Association of the platelet PLA polymorphism of
glycoprotein IIb/IIIa and the fibrinogen Bb448 polymorphism
with myocardial infarction and the extent of coronary artery
disease. Circulation 1997; 96: 142-143.
37. Zotz RB, Winkelmann BR, Namk M, Giers
G, Maruhn Debowski B, Marz W, et al. Polymorphism of
platelet glycoprotein IIIa: human platelet antigen 1b (HPA-1b/
PLA2) is an inherited risk factor for premature myocardial
infarction in coronary artery disease. Thromb Haemost 1998;
79: 731-735.
38. Herrmann SM, Poirier O, Marques-Vidal,
Evans A, Arveiler D, Luc G, et al. The Leu33/ Pro
polymorphism (PLA1/ PLA2) of the glycoprotein IIIa (GPIIIa)
receptor is not related to myocardial infarction in the ECTIM
study. Thromb Haemost 1997; 77: 1179-1181.
39. Bottiger C, Kastrati A, Koch W, Mehilli
J, Seidl H, Schomig K, et al. HPA-1 and HPA-3
polymorphisms of the platelet fibrinogen receptor and coronary
artery disease and myocardial infarction. Thromb Haemost 2000;
83: 559-562.
40. Feng D, Lindpaintner K, Larson MG, Rao
VS, O’Donnell CJ, Lipinska I, et al. Increased platelet
aggregability associated with platelet GPIIIa PLA2
polymorphism. The Framingham Offspring Study. Arterioscler
Thromb Vasc Biol 1999; 19: 1142-1147.
41. Lasne D, Krenn M, Pingault V, Arnaud E, Fiessinger J,
Aiach M, et al. Interdonor variability of platelet
response to thrombin receptor activation: influence of PLA2
polymorphism. Br J Haematol 1997; 99: 801- 807.