|
|
Case Reports Indian Pediatrics 2001; 38: 919-922 |
||
|
Carbohydrate-Deficient Glycoprotien Syndrome (CDGS) 1a |
||
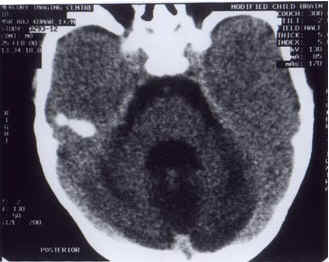
Jaeken and his colleagues in 1980 described, for the first time, two 19 months twin girls with psychomotor retardation, cerebellar hypoplasia and abnormalities of several unrelated glycoproteins(1). Subsequently defi-ciency of carbohydrate moieties of the glyco-proteins was identified as the underlying biochemical defect and the disorder name as carbohydrate-deficient glycoprotein syndrome (CDGS)(2). Similar cases have been reported from all over the world. However, because of the paucity of such reports in the Indian litera-ture, we are documenting our observations. Case Report A 1½-year-old boy presented with global developmental delay, failure to thrive and episodic vomiting since birth. There was no history of seizures, episodes of unconscious-ness, neurodeficits, recurrent infections or jaundice. He was born to nonconsanguineous parents and his elder sister had died of similar illness at 1 year of age. Examination revealed a malnourished child with weight 6.9 kg (less than 5th percentile) and head circumference 42.0 cm (less than 5th percentile). He had abnormal facies consisting of broad nasal bridge, slightly prominent mandible and excessive growth of hair on forehead with synophrys. Open anterior fontanel, bilateral cryptorchidism and inverted nipples were also noted. His skin had a peculiar thick and sticky feel and a peau d’ orange look. Abnormal deposits of fat were seen in the gluteal, pubic and supragluteal areas (Fig. 1). A few subcuta-neous nodules were present on the dorsal aspect of toes. Examination of cardiovascular and respiratory systems did not reveal any abnormalities. No hepatosplenomegaly was found on abdominal examination. Neuro-logical examination revealed esotropia, hypo-tonia and areflexia. Fundus examination was normal. Rest of the examination was un-remarkable except for limited extension of knee and elbow joints. Hemogram, liver and kidney functions, coagulogram, blood glucose and abdominal ultrasound were all normal. CT scan of head revealed ponto-cerebellar hypoplasia (Fig. 2). Child developed one episode of vomiting and drowsiness during the hospital stay but recovered within 48 hours of symptomatic treatment. Discussion Carbohydrate-deficient glycoprotein syn-drome is a newly recognized group of inherited multisystemic disorders associated with abnormal glycosylation of a number of serum glycoproteins(3,4). Following its first descrip-tion by Jaeken and colleagues in 1980(1), several cases have appeared in literature. Originally described from Belgium, Sweden and Norway, the syndrome is being identified from other parts of the world. The present case, to the best of our knowledge, is the first to be described from India. Four subtypes of CDGS have been delineated on the basis of clinical presentation and biochemical changes of glycosylation of serum transferrin. Enzyme deficiencies have been characterized for type Ia, Ib, Ic and II(3,4). The commonest variety of CDGS is type Ia. About 300 cases have been described so far(1). In the most instances, a deficiency of phosphomannomutase (PMM) has been found. The inheritance is autosomal recessive and gene responsible for the disorder has been mapped to chromosome 16p, though genetic heterogeneity has been seen(3,4). Other subtypes of CDGS (type Ib, Ie, II, III, and IV) have been found to be very rare and fewer than 10 patients have been described in each of them(3). Clinical manifestations of CDGS Ia are quite characteristic and show an age dependent expression. Neurological dysfunction is present in all, with psychomotor retardation, failure to thrive, hypotonia, hyporeflexia and acquired microcephaly. Seizures, unexplained stuporous or stroke-like episodes occur in some cases. Infratentorial, particularly cerebellar hypoplasia is a frequent patho-radiological finding. Most of these findings were observed in our case. Extraneurological involvement is variable and includes alarming episodes of liver failure, nephritic illness, pericardial effusion, cardiac tamponade, severe infections and complex coagulopathies. Esotropia is the most common ocular finding. Retinitis pigmentosa is a late complication of the disorder(3-6). Majority of the children have typical dysmorphic features. Facies are characterized by esotropia, high nasal bridge, prominent jaw and large external ears. Most characteristic, however, are the lipocutaneous manifestations including areas of lipohypertrophy producing pads of fat around buttocks, supragluteal region and pubic area. These are usually present at birth. The skin has a peculiar thick and sticky feel and a peau d’ orange appearance, especially on the extremities. Some patients develop lipoatrophic changes on the lower limbs. Additional dysmorphic features include inverted nipples (almost a universal finding), restricted joint mobility and cryptorchidism (3,5,7). A clinical diagnosis of CDGS Ia can usually be made from these characteristic dysmorphic features especially the lipocutaneous changes, in association with psychomotor retardation and the neuro-radiological findings(4,7).
Fig. 1. Photograph showing gluteal and supragluteal pads of fat.
Fig. 2. CT scan head showing hypoplasia of infratentorial structures particularly cerebellum and pons. Biochemical hallmark of CDGS is hypoglycosylation of serum glycoproteins. Serum transferrin shows most pronounced changes, which can be determined quali-tatively by immuno-isoelectric focusing or quantitatively by estimating carbohydrate-deficient transferrin (CDT) by anion-exchange chromatography(8). False positives can occur in patients with galactosemia, fructosemia or chronic alcoholism(4). Other serum glyco-proteins like antithrombin-III and thyroid binding globulin can be measured to provide additional support for the diagnosis(3). The final confirmation however rests on enzyme assay or mutational analysis. We did not have access to these sophisticated investigations. The development expression of CDGS la is striking(3-5). Illness during infancy is characterized by life threatening episodes of liver, heart or multiorgan failure or severe infections resulting in considerable morbidity and mortality. Seizures and stroke like episodes are frequent in early childhood and often occur in association with intercurrent febrile illnesses. Course appears to stabilize later but peripheral neuropathy and retinitis pigmentosa become more prominent. Survival to adulthood is accompanied by hypogonadism, skeletal disproportions, severe ataxia and stable mental retardation(3-5). No effective treatment is available for CDGS la. Prenatal diagnosis of CDGS la has been made through a combina-tion of enzymatic and genetic linkage analysis and can be offered to the affected family(9). The diagnosis of CDGS la should be considered in any child with failure to thrive, developmental retardation, unexplained organ failure, stupor or stroke like episodes, lipo-cutaneous changes and the neuroradiological signs, i.e., cerebellar hypoplasia(3,4). It is likely that CDGS has a wider spectrum than what is currently known. Increasing the use of screening tests (qualitative or quantitative transferrin assay) in children presenting with otherwise unexplained isolated symptoms and signs of CDGS would help define the limits of the disorder. Contributors: JSG, SM and SB were involved in literature search and preparation of the manuscript. BP and VRP co-drafted and critically evaluated the paper. JSG will act as the guarantor of the paper.
Funding: None.
|
![]()