|
Rashmi Kapoor
P.M. Gadre
Pradeep Mattoo
Rajeev Agrawal
From the Departments of Pediatrics and Orthopedics, Regency Hospital Ltd., Kanpur, India.
Reprint requests: Dr. Rashmi Kapoor, Regency Hospital Ltd., A-2, Sarvodaya Nagar,
Kanpur 208 005, India.
Manuscript Received: October 22, 1997; Initial review completed: December 10, 1997; Revision Accepted: January 20, 1998
Fibrodysplasia ossificans progressiva (FOP) also known as myositis ossificans progressiva is a rare inherited connective tissue disorder characterized by the heterotopic development of bone in areas of body where bone is not usually present such as ligaments, tendons and
muscles(1).
The disease is extremely rare and fewer than hundred adult cases have been re- ported in USA and UK(2). We came across a typical case of FOP in a 21/2 years old child with extensive involvement. To the best of our knowledge, a case of full fledged
"FOP" at 21/2 years of age has not been reported.
Case Report
A 21/2-year-old female child, third in birth order, having three asymptomatic and apparently normal siblings, presented to us with multiple, hard, painless swellings over neck and back of 11/2 years duration, inability to move neck of 1 year duration and inability to move right arm at elbow of 6 months duration. There was no
history of trauma, fever, bleeding tendencies, swelling of joints or rash. No similar ailment was present in any Other family member of the child. The child on examination had grade I Protein energy malnutrition and mild pallor. The examination of the musculo skeletal system revealed multiple, bony-hard, non tender and immobile swellings scattered over occipital area of scalp, neck and back (Fig. 1). The child had gross restriction of the movements of neck
due to ossified neck muscles and also gross restriction of movements at right elbow due to the ossification of the muscles around the joint especially the biceps muscle. She had short and deformed big toes of both feet. Rest of the systemic examination was unremarkable. Investigations revealed mild microcytic hypochromic anemia while other hematological and biochemical parameters were: TLC- 13,600/cu mm (P40, L49, ElO, M1), serum Calcium-8.8
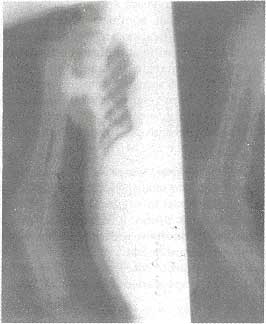
mg/dl; serum Phosphorus-5.9 mg/dl; CPK-77 IU/L, SGPT -22 IU/ L; and alkaline phosphatase-10 KAU. The radiological skeletal survey revealed heterotopic ossification of muscles at multiple sites (muscles of shoulder girdle, biceps, triceps and paraspinal region) without any other significant bone deformity (Fig. 2). The child was given supportive therapy after properly explaining the nature of ailment to her parents.
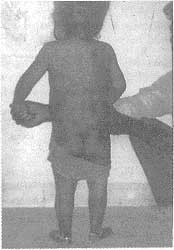 |
Fig. 1. Photograph showing multiple swellings over the back. |
 |
|
Fig. 2. X-ray right arm (AP and lateral views) showing ossification of muscles at multiple
sites.
|
Discussion
FOP is an extremely rare disorder
characterized by a physical handicap due to intermittently progressive heterotopic ossification involving muscles, ligaments and tendons. Major signs include skeletal malformations and/or abnormally short and malformed toes and fingers(3). The heterotopic development of bone leads to stiffness in affected areas and may also limit movement of affected joints. Scalp baldness, deafness and
and mental retardation may be other associated features(4).
While normal serum calcium and phosphate levels ruled out metastatic calcification, the typical skeletal malformations and radiologically proved heterotopic ossification ruled out dystrophic calcification (for example, dermatomyositis) as a cause for the clinical presentation in our case.
The average reported age at onset of ossification is 5 years (range, birth to 25 years). The most common sites of early heterotopic ossification ,are the neck, spine and shoulder girdle and almost all patients have a severely restricted mobility of arms by the age of 15 years. Premature death of. ten results from respiratory failure due to fixation of thoracic cage(5). In our case, the child had heterotopic ossification of all these sites at the age of 21/2 years.
Although most cases of FOP are sporadic, sufficient examples of affected twirls and triplets exist
to suggest a genetic basis. The disease is probably determined as an autosomal
dominant trait which has complete penetrance but variable expressivity(3). Sporadic cases are explained by high level of spontaneous mutations(2).
The biopsy of a lesion in FOP reveals
normal endochondral osteogenesis at heterotopic sites. The association of post- natal heterotopic endochondral osteogenesis with congenital malformation of the great toes suggests that FOP is a disorder of defective induction of endochondral osteogenesis. However, a lesional biopsy is seldom required to make the diagnosis and, in fact, it has been shown that it may exacerbate the condition(6).
The rarity of the disease limits the understanding of its exact pathogenesis and no treatment is as yet available to control or cure the disease process.
|
1.
McKusick VA. Fibrodysplasia ossificans progressiva. In: Heritable Disorders of Connective Tissue St. Louis, C.V. Mosby 1993; pp 501-518.
2.
Connor JM, Evans DAP. Genetic aspects of fibrodysplasia ossificans progressiva.
J
Med Genet 1982; 19: 35-39.
3.
Connor JM, Evans DAP. Fibrodysplasia ossificans progressiva: The clinical features and
natural history of 34 patients.
J
Bone Joint Surg 1992; 64: 76-83.
4.
Becker PE, Yon Knorre GV. Myositis Ossificans Progressiva. Ergeb. Inn:. Med Kinderheilk 1968; 27: 1-31.
5.
Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, et al. The nahiral
history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva:
A study of forty four patients.
J
Bone Joint Sutg. 1993; 75A: 215-219.
6.
Kaplan FS, Tabas JA, Gannon FH. The histopathology of fibroqysplasia ossificans progressiva: An endochondral
process.
J
Bone Joint Surg 1993; 75A: 220-
230.
|
