S.K. Chowdhary
K.L. Narasimhan
R. Ray
R.K. Marwaha
S.K. Mitra
From the Departments of Pediatric Surgery,
Pathology and Pediatrics, Post Graduate Institute of Medical
Education and Research, Chandigarh 160 012, India.
Reprint requests: Dr. S.K. Chowdhary,
Assistant Professor, Department of Pediatric Surgery, PGIMER,
Chandigarh 160012, India.
E-mal: sujitchowdhary@hotmail.com
Manuscript Received: February 12, 1999;
Initial review completed: March 24, 1999;
Revision Accepted: September 22, 1999
Epitheloid sarcoma is a soft tissue tumor of
young adults. A majority of them arise in the subcutaneous tissue
or dermis of the upper extremity around the hand and forearm and
start as a small nodule. They have a propensity to recur locally
after excision and in a few cases may metastasise. Although this
tumor has been reported in children, we have not come across
description of this tumour in a neonate.
A one-month-old boy was brought with a lump in
the right hand, which had gradually grown since birth (Fig. 1).
This was a multi-nodular swelling measuring 8 ´
10 cm arising from the palmar aspect of the right hand extending
between the proximal palmar crease and web spaces. The overlying
skin had characteristic neovascularisation and was fixed to
underlying structures. The right axilla had a solitary lymph node
3 ´
3cm, and there was, in addition a nodule in the epigastrium.
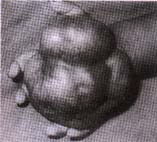
Fig.
1. Photograph demonstrating the tumor arising from the palmar
aspect of the right hand with typical neovascularisation.
An incisional biopsy of the mass in the right
hand revealed nodular arrangement of spindle and epitheloid cells
with densely staining large nuclei (Fig. 2).
Immunohistochemistry of the specimen revealed positive staining
for vimentin and negative staining for desmin. Keratin staining
could not be done. Aspiration cytology of the nodular lump in the
axilla and epigas-trium revealed similar spindle and epitheloid
cells.
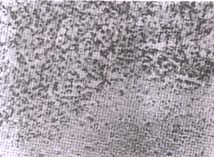
Fig. 2. Histological section from the tumor
showing epitheloid and spindle cells with large densely
staining
nuclei.
The parents were not in favour of surgery.
Adjuvant chemotherapy was started with weekly doses of vincristine,
cyclophosphamide and actinomycin D. However, the tumor continued
to grow and three weeks later the baby died with febrile
neutropenia.
Benign and malignant soft tissue tumors are not
uncommon in children. Infantile fibro-matosis and fibrosarcoma
together constitute more than two thirds of the neonatal solft
tissue tumors(1). These tumors frequently start as nodule in the
extremity and show characteristic neovascularisation. Around 75%
of cases recur locally and upto 40% can metastasise with fatal
results(2). These nodules are often mistaken as an inflammatory
nodule resulting in a delay in treatment. The nomenclature of the
tumor is derived from the histological appearance of epithelium
like cells merging imperceptibly with spindle cells with minimal
cytological pleomor-phism, prominent nuclei and characteristic
necrosis in the center of nodular arrangement of cells. A peculiar
variant of synovial sarcoma was described in literature
characterized by polygonal and polyhedral cells bearing striking
resemblance to epithelium long ago(3). The tumor tissue has
striking acidophilic appearance due to staining characteristic of
cytoplasm and dense mass of hyalinized collagen forming the stroma
of the tumor. Calcification may be seen in the area of central
necrosis in a few cases. Immuno histochemically, there is
positivity for vimentin, keratin, epithelial membrane antigen, CD
34, and tissue polypeptide antigen. How-ever, the co-expression of
vimentin and keratin is thought to be a characteristic of this
tumor. The exact histogenesis of the tumor remains obscure,
although it does demonstrate attempt at epithelial
differentiation(4). The tumor is known to spread to noncontiguous
areas of the skin, soft tissue, fascia and bone as well as by
direct extension. A more aggressive clinical course is associated
with a proximal or axial location, increased size and depth,
hemorrhage, increased mitotic figures, rhabdoid features and
vascular invasion.
Gross et al. reviewed 8 children with
epitheloid sarcoma, two of whom had meta-stases and both were dead
within 1 year(5). de Vries earlier reported another series on
children with epitheloid sarcoma with similar fatal result in 50%
cases(6).
The highly malignant nature of the tumor is
well documented. A high index of suspicion and early biopsy will
promote early diagnosis. Local excision alone fails in a majority
of the patients. Radical excision or limited amputation is the
surgical treatment of choice. Adjuvant radiotherapy and
chemotherapy have a limited role.
1. Stevens MCG. Neonatal tumors. Arch Dis Child
1988; 63: 1122-1125.
2. Chase DR, Enzinger FM. Epitheloid sarcoma:
Diagnosis, prognostic indicators and treatment. Am J Surg Pathol
1985; 9: 241-263.
3. Enzinger FM. Epitheloid sarcoma: A sarcoma
simulating a granuloma or a carcinoma. Cancer 1970; 26: 1029-1041.
4. Chase DR, Enzinger FM, Weiss SW, Langloss
JM.
Keratin in epitheloid sarcoma: An immuno-histochemical study. Am J
Surg Pathol 1984; 8: 435-441.
5. Gross E, Bhaskar NR, Pappo A, Bowman L,
Shearer P, Kaste P, et al. Epitheloid sarcoma in children.
J Pediatr Surg 1996; 31: 1663-1665.
6. de Vries J, Hoekstra HJ, Oosterhuis JW, Postma A,
Schraffordt Koops H. Epitheloid sarcoma in children and
adolescents: A report of four cases. J Pediatr Surg 1989; 24:
186-188.
|
