|
clinicopathological conference |
|
|
Indian Pediatr 2020;57:
349-355 |
 |
An Infant with Severe Anemia and
Hypoalbuminemia
|
|
Jogender Kumar1, Debajyoti Chatterjee2, Sadhna
B Lal3 and Praveen Kumar1
From
1Departments of Pediatrics, 2Histopathology and 3Gastroenterology,
Post Graduate Institute of Medical Education and Research, Chandigarh,
India.
Correspondence to: Dr Jogender Kumar, Assistant Professor,
Department of Pediatrics, Post Graduate Institute of Medical Education
and Research, Chandigarh 160 012,India.
Email:
[email protected]
|
|
We discuss the case of a two-month-old girl admitted with complaints of
progressive pallor, generalized body swelling and pale colored stool
since the neonatal period. On examination, severe pallor, chubby cheeks
and moderate hepatomegaly were noted. Investigations revealed isolated
anemia, transaminitis, conjugated hyperbilirubinemia, prolonged
prothrombin time and hyperlipidemia. She died due to severe sepsis,
shock, and pulmonary hemorrhage. An autopsy revealed characteristic
histopathology findings of cystic fibrosis in the liver, lungs, and
pancreas. Genetic analysis performed on autopsy tissue was positive for
F508del compound heterozygous (WT/F508del) mutation, confirming the
diagnosis of cystic fibrosis.
Keywords: Autopsy,
Cholestatic jaundice, Cystic Fibrosis.
|
|
CLINICAL PROTOCOL
History: A
two-month-old girl, a product of non-consanguineous marriage, presented
with complaints of gradually increasing generalized body swelling
starting from the age of 15-20 days. It was associated with progressive
pallor, for which she had received one packed red blood cell (PRBC)
transfusion at 1½ month of age. Since one month of age, she had passed
8-10 pale-colored semisolid stools per day. She also had fast breathing
for 15 days prior to admission. There was no history of jaundice, high
colored urine, bleeding from any site, mucus in stool, lethargy, poor
feeding, irritability, seizures, and encephalopathy. She was born at
term gestation with a birthweight of 2.3 kg and was admitted in the
intensive care unit for five days in view of abdominal distension and
respiratory distress since birth. She passed meconium at the end of day
2 of life, following which abdominal distension resolved. The baby was
exclusively breastfed, immunized for age and developmentally normal for
age. The elder sibling had a tracheo-esophageal fistula and had died on
day two of life.
Clinical examination: At admission, she was
alert and active with a heart rate of 128/min, respiratory rate of
58/min, good volume pulses, normal capillary refill time, and 100%
oxygen saturation on room air. She had severe pallor and anasarca. She
weighed 3.5 kg (-4.28 Z) and the occipitofrontal circumference was 34.1
cm (-4.51 Z). The baby had very prominent chubby cheeks. Respiratory
system examination showed bilateral basal crepitations. The abdomen was
distended with liver being palpable 6 cm below right costal margin and 3
cm below the left costal margin in the midclavicular line (span 9-10
cm), soft-to-firm in consistency, non-tender, with ill-defined borders.
The spleen was not palpable. Examination of cardiovascular system and
central nervous system was normal. Fundus examination did not show any
chorioretinitis or cherry red spot.
Laboratory investigations:
She had normocytic normochromic anemia (haemoglobin 6.5 g/dL),
leuko-cytosis (white cell count 34,360/µL, differential counts (53%
polymorphs), normal platelet count, transaminitis (alanine
aminotransferase – 79 IU/L and aspartate aminotransferase – 367 IU/L),
cholestatic jaundice (total bilirubin – 4 mg/dL, direct – 3.3 mg/dL),
severe hypoalbuminemia (1.5 g/dL), deranged coagulation profile
(prothrombin time – 20.9 seconds, activated thromboplastin time – 44.6
seconds) and hyperlipidemia (total cholesterol – 255 mg/dL, triglyceride
– 289 mg/dL) and a high C-reactive protein (33.6 mg/L). Arterial blood
gas analysis revealed respiratory alkalosis. She also had persistent
hyponatremia (126 mEq/L) and hypochloremia (93 mEq/L). Chest radiograph,
stool examination, immunoglobulin profile (IgG- 346 mg/dl, IgA- 51
mg/dL), and T-cell subset assay (CD3+= 55.43%, CD 19+= 32.34%,
CD56+=4.84%, CD3+ CD56+= 0.46%) were normal. Blood sugar was 96 mg/dL
and serum ammonia was 225.8 µmol/L. Urinary aminoacidogram could not be
done. Human immunodeficiency virus, cytomegalovirus, and toxoplasma
serology were negative. Ultrasound abdomen showed hepatomegaly with
normal liver echotexture. The cranial ultrasound did not show any
structural malformation or calcification. The blood culture sent at
presentation was sterile; however, repeat blood culture sent on day 4
grew Staphylococcus hominis (sensitive to ciprofloxacin, clindamycin,
teicoplanin and vancomycin; oxacillin and erythromycin).
Course
management: The infant received intravenous cefotaxime, cholestatic
regimen [1] and PRBC transfusion. On day 3 of hospital stay, she had one
episode of fresh blood in stool along with deranged coagulation profile;
therefore, fresh frozen plasma was transfused. However, on the next day
she worsened further in the form of tachycardia, poor pulses, and
prolonged capillary refill time, for which antibiotics were empirically
upgraded to vancomycin, meropenem and amphotericin B. Fluid bolus and
inotropic support was given for the shock. On day 5 of hospital stay,
she had further cardiorespiratory worsening for which she was intubated
and kept on manual intermittent positive pressure respiration. On the
same day she developed hypocalcemia and hypokalemia requiring
correction. She deteriorated further and received intravenous
immunoglobulin, multiple fluid boluses and inotropes (dopamine,
dobutamine, and adrenaline). However, she had worsening of shock and
hypoxemia followed by massive pulmonary haemorrhage leading to cardiac
arrest and death on day 5 of hospital stay.
Unit’s final
diagnosis: Glycogen storage disorder with refractory septic shock and
pulmonary hemorrhage.
DISCUSSION
Clinical
discussant: We have a two-month-old girl presenting with severe pallor,
anasarca, pale stool, chubby cheeks, failure to thrive, microcephaly,
moderate hepatomegaly, transaminitis, cholestatic jaundice, and
hyperlipidemia. She had isolated normocytic normochromic anemia, with
deranged coagulation profile and required two blood transfusions during
the initial two months of life. This case can be analyzed with respect
to underlying disease and the pre-terminal events. The primary analysis
suggests multi-system disease with predominant hepatic involvement.
There are many causes of isolated hepatomegaly with the above findings.
Of these, intrauterine infections (TORCH), hemophagocytic syndrome and
metabolic/storage disorders can present like this child. Intrauterine
infections (particularly CMV and toxoplasma) are unlikely in the absence
of splenomegaly, thrombocytopenia, chorioretinitis, hepatic, and
cerebral calcification. Moreover, the serology for CMV and toxoplasma
was negative. Hemophagocytic syndrome is also unlikely in the absence of
fever, splenomegaly, and bicytopenia. The NK cell activity was also
normal. The possible storage/metabolic disorders can be glycogen storage
disorder (GSD), lipid storage disorder (Gaucher and Niemann-Pick
disease), iron storage (neonatal hemochromatosis due to gestational
alloimmune liver disease, GALD), alpha-1 antitrypsin deficiency,
galactosemia, cystic fibrosis (CF), and citrin deficiency. However, in
the absence of splenomegaly, thrombocytopenia, cardiac malformation, and
cherry-red spot; lipid storage disorders are less likely. GALD is less
likely as it starts from fetal life itself and frequently present in the
early neonatal period with prematurity, acute hepatic failure, very high
bilirubin, hydrops, and renal failure. Alpha -1 antitrypsin deficiency
can have similar presentation, but lack of respiratory symptoms,
splenomegaly, and chubby cheeks make it less likely. Presence of chubby
cheeks and lack of significant coagulopathy are against galactosemia.
The rest of the metabolic disorders (GSD, citrin deficiency and CF) are
strong possibilities. Among GSDs; type I, III, VI, and IX have a
predominant hepatic presentation with moderate hepatomegaly, but only
GSD type I can lead to severe anemia at this age. However, the presence
of significant microcephaly, severe hypoalbuminemia, prolonged PT/APTT
and lack of hypoglycemia even in extreme sickness is against GSD type I.
Citrin deficiency has a wide spectrum and can present in infancy
as Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD)
and/or Failure to thrive and dyslipidemia caused by citrin deficiency
(FTTDCD) [2]. The classical features of citrin deficiency are low birth
weight, intrauterine growth restriction, microcephaly, chubby cheeks due
to excessive lipid deposition, hepatomegaly, neonatal cholestasis,
features of liver failure and hemolytic anemia. They also have mild
hyperammonemia, hyperlipidemia, increased alpha-fetoprotein, and fatty
liver. Diagnosis is based on abnormal newborn screen (aminoaciduria)
followed by genetic analysis. All the features in the index case are
consistent with the citrin deficiency except, normocytic anemia,
hyponatremia, hypochloremia, and pulmonary symptoms. However, chubby
cheeks with predominant hepatic manifestations are classical of GSD and
citrin deficiency. Since GSD is less likely in this case, citrin
deficiency may be considered as a strong differential diagnosis.
Cystic fibrosis-associated liver disease (CFLD) can present in early
infancy with features of hepatomegaly, cholestasis, transaminitis,
dyslipidemia, and severe anemia along with hypoalbuminemia. Severe
anemia can be the first sign of cystic fibrosis in 7-10 % of infants
with cystic fibrosis and the concomitant presence of severe
hypoalbuminemia makes it more likely [3]. The anemia of CF is normocytic
normochromic and may precede several months of respiratory symptoms. The
index case also has delayed passage of meconium along with hyponatremia
and hypochloremia, further favoring the diagnosis of CF. However, the
presence of chubby cheeks and normal liver echotexture is not consistent
with CF. Overall, citrin deficiency and CF are the most likely clinical
differential diagnosis in the index case. The prominence of chubby
cheeks strongly favors citrin deficiency. However, disregarding chubby
cheeks, the clinical picture is consistent with CF.
In terminal
stages, the infant had severe sepsis (most likely of bacterial origin)
characterized by increasing CRP, high leukocyte count, and decreasing
platelets. She developed refractory septic shock leading to multiorgan
dysfunction and subsequently died.
Chairperson: As per the
clinical experience of the unit; which is most common among GSD, Citrin
deficiency, and CF?
Pediatric gastroenterologist 1: During
hospitalisation, GSD was our first possibility. However, on
retrospective analysis of the biochemical profile (AST much higher than
ALT, dyslipidemia, lack of hypoglycemia and lactic acidosis) in the
presence of very prominent chubby cheeks developing over two months,
citrin deficiency seems more likely. Citrin deficiency has been
described over the last few years, primarily from Japan and South East
Asia. It is perceived to be rare, though not uncommon. However, it is
unlikely that this case will have a definite histopathological picture
of citrin deficiency. In our experience, GSD is the most common clinical
condition. However, recently we were able to diagnose a few cases of
citrin deficiency too. The children with citrin deficiency generally do
well and improve over a period of time. This child died because of
sepsis, not of citrin deficiency per se.
Pediatric pulmonologist:
There was delayed passage of meconium with some component of diarrhea.
This child is a classic picture of cystic fibrosis. The chubby cheeks
may be due to hypoalbuminemia and edema which may give a false
impression of good nourishment.
Neonatologist: In CF, we expect a
low level of cholesterol, whereas the cholesterol was significantly
elevated in this case.
Pediatric Gastroenterologist 1:
Galactosemia is a mimicker of citrin deficiency. However, the chubby
cheeks and lack of significant coagulopathy are strongly against it.
Pediatric Neurologist 1: Niemann pick type C can present like
hydrops fetalis in early infancy. Tyrosinemia can also have a similar
presentation.
Pediatric Gastroenterologist 2: Lack of
splenomegaly and thrombocytopenia are against Niemann pick type C. In
tyrosinemia, the prominent features are severe coagulopathy and
fulminant hepatic failure, which was not seen in this case. I would like
to keep the possibility of a congenital disorder of glycosylation (CDG)
type Ib/Ih.
Pediatric Neurologist 2: Severe anemia at an early
age is not usual in storage disorders. Therefore, it must be due to the
marrow involvement and Pearson marrow-pancreas syndrome can be
considered as a differential diagnosis. CDG also has similar features
along with abnormal fat distribution.
PATHOLOGY PROTOCOL
A complete autopsy was performed in the index case. There was 50 ml
straw-coloured fluid in the pleural and peritoneal cavity.
Liver
weighed 226 g and was slightly enlarged, soft, and bile stained
(Fig. 1). Gall bladder was normal in size and the extrahepatic
biliary tree was patent. Microscopically, the liver showed expansion of
the portal tracts with mild fibrosis and extensive bile ductular
proliferation. There was a focal porto-portal bridging, producing focal
biliary cirrhosis. There was extensive macrovesicular steatosis, along
with intrahepatocytic and intracanalicular cholestasis, producing
feathery degeneration of the hepatocytes (Fig. 1 b-d).
However, there were no PAS-positive diastase resistant inclusions in the
hepatocytes. Larger bile ducts and bile ducts at porta hepatis were
markedly dilated and filled with inspissated secretions. These
inspissated secretions were Periodic-Acid-Schiff (PAS) positive,
diastase-resistant and strongly positive for the alcian blue, indicating
mucinous nature. The biliary epithelium showed evidence of mucinous
metaplasia. Similar inspissated secretions were also seen in peribiliary
glands (Fig. 1 e-f). The pancreas was firm in
consistency. It showed marked dilatation of the ducts filled with
inspissated secretions. There was marked intra and inter-lobular
fibrosis with loss of acini, and focal lymphomononuclear infiltrate
(Fig. 2 a-c). Spleen (12 g) showed normal white and red
pulp. Stomach, esophagus, small intestine, and large intestine were
grossly unremarkable. There were no inspissated secretions. Perl stain
did not reveal any evidence of excess iron deposition in liver, spleen
or pancreas.
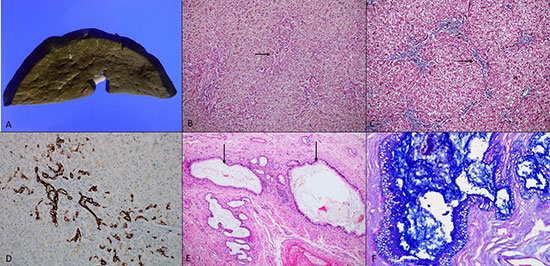 |
| Fig. 1 (a) Gross
photograph of liver shows extensive bile staining.(b)
Liver shows irregular portal tracts (black arrow), with
macrovesicular steatosis and cholestasis (hematoxylin
and eosin, ×100). (c) Masson’s trichrome highlights
irregular portal fibrosis with occasional porto portal
bridging (black arrow) (×100). (d) Bile ductular
proliferation is highlighted by cytokeratin 7
(immunohistochemistry, ×200). (e) Section from porta of
liver shows marked dilatation of larger bile ducts,
filled with inspissated secretions (black arrow)
(hematoxylin and eosin, ×100). (f) Alcian blue- Periodic
acid Schiff stain highlights the inspissated secretions
and mucinous metaplasia of the biliary epithelium
(×400). |
Lung weighed 95 g and the bilateral
pleura were dull. Bilateral lower lobes were consolidated. There
was extensive hemorrhagic discoloration of both lungs (left >
right). Sections of the lungs showed dilated bronchioles, filled
with inspissated secretions. Inspissated secretions were also
seen in the main bronchus (Fig. 2 d-f). The
subepithelial glands were hypertrophied and showed inspissated
secretions. In addition, lungs showed exuberant capillary
proliferation in the alveolar septa, with the capillaries
infiltrating the wall of the pulmonary arteries, producing
pulmonary capillary hemangio-matosis. Extensive fresh pulmonary
hemorrhage was noted. Also, there was evidence of
bronchopneumonia with diffuse alveolar damage.
 |
| Fig. 2 (a) Pancreas
shows dilated ducts with inspissated secretions (black
arrows) and parenchymal fibrosis (hematoxylin and eosin,
×100). (b) Alcian blue- Periodic acid Schiff stain
highlights the inspissated secretions in the pancreatic
ducts (black arrows) (×100). (c) Masson’s trichrome
stain highlights inter and intralobular fibrosis in
pancreas (black arrows) (×100). (d) Section from lung
shows dilated bronchioles filled with inspissated
secretions (black arrows) (hematoxylin and eosin, x100),
which is highlighted by (e) periodic acid Schiff (×100)
and (f) Alcian blue stain (×100). |
Heart (25 g) showed normal chambers
and valves. Kidneys (46 g) showed normal fetal lobulations.
Microscopic examination did not reveal any pathology. Brain (491
g) showed normal sulci and gyri. No gross or microscopic
pathology was seen. Bone marrow was normocellular for age and
showed marked erythroid hyperplasia. Other hematopoietic
lineages were adequately represented. Thymus showed extensive
cystic degeneration of the Hassel corpuscles likely due to
stress-induced involution.
Genetic mutations in the CFTR
gene: Peripheral blood was collected at autopsy and was
subjected to CFTR gene mutation by the mass array. A limited
CFTR gene panel was examined and showed F508del compound
heterozygous (WT/F508del) mutation. Mass array performed for FIC
2 and 3 genes did not reveal any mutation.
Final autopsy
diagnosis:
• Cystic fibrosis involving lung, pancreas,
and liver (F508del compound heterozygous)
• Bronchopneumonia
with diffuse alveolar damage
• Pulmonary capillary
hemangiomatosis
• Pulmonary hemorrhage
• Erythroid
hyperplasia of bone marrow
Pediatric Pulmonologist: The
histopathology shown here is the classical book picture of
cystic fibrosis. This mutation is likely a compound heterozygote
and they do not have a very good phenotypic-genotypic
correlation. The parents should also be evaluated for the
mutation. If we would have suspected in the antemortem period,
the course would have been different and treatment can be
offered in earlier stages.
Pediatric Gastroenterologist
1: The clinical picture was not very classical of CF. Even if we
would have done an antemortem percutaneous liver biopsy, the
conclusive diagnosis was unlikely. The focal biliary cirrhosis
is a very nonspecific finding and is seen in myriad conditions.
Even sweat chloride testing is not feasible at this age.
Therefore, it is very difficult to make an antemortem diagnosis
of CF at such an early age. The classical features appear during
adolescence only.
Pathologist 1: The pulmonary capillary
hyperplasia shown in histopathology is a reactive finding and is
commonly observed during infancy. It should not be confused with
the pulmonary capillary hemangiomatosis (PCH), which is a
diagnosis of exclusion. PCH should show infiltration of pleura,
septa, veins, and the vessel wall.Moreover, in PCH, the
capillaries form a nodule and show multiple layers. Therefore,
here it was just a reactive pulmonary capillary hyperplasia.
Pediatric Pulmonologist: PCH is also known to occur in CF
when they develop pulmonary artery hypertension. There is
hypersecretion of VEGF that leads to this finding in CF.
Clinical discussant: Everything was consistent with CF, but the
chubby cheeks took us away from it; likely these were a
manifestation of severe hypoalbuminemia.
Pathology
discussant: The histopathology shown here is very characteristic
of reactive pulmonary capillary hemangiomatosis, which can be
associated with cystic fibrosis and various metabolic liver
diseases.
Pathologist 2: Even if we would have done an
antemortem liver biopsy, the diagnosis was unlikely. Here we
have a whole liver, so we could show classical findings. In
suspected CF, we should always look for changes in Brunner gland
in the duodenum.
Pathologist 3: Liver biopsy plays an
important role in the work-up of infantile cholestasis.
Although, it may not be diagnostic in all cases, it provides
important information to exclude other conditions presenting as
infantile cholestasis such as congenital hepatic fibrosis,
extrahepatic biliary atresia, progressive familial intrahepatic
cholestasis or paucity of intrahepatic bile duct.
DISCUSSION
Cystic fibrosis is one of the
commonest life-limiting autosomal recessive monogenic disorders.
Initially thought to be affecting the Caucasians only, its
presence is pan-ethnic [4]. It is caused by mutations in
the CFTR (cystic fibrosis transmembrane conductance regulator)
gene. Till now, more than 1500 mutations have been described in
CF, of which the deletion of phenylalanine at codon 508 (dF508)
is the commonest. Different mutations have varying genotypic
effects on CFTR function and can result in different phenotypic
expression of the disease [5,6]. The manifestations of CF may
begin in early infancy itself in the form of delayed passage of
meconium, meconium ileus, recurrent loose stool, malabsorption,
cholestatic jaundice and failure to thrive. Later they have
recurrent respiratory infections, features of malabsorption and
involvement of many other organ systems, namely endocrine,
hepatobiliary and reproductive system [7].
CFLD, the
liver involvement in cystic fibrosis can be found in infancy in
13%-17% cases of cystic fibrosis [8,9]. The presentation of CFLD
may vary from asymptomatic transaminitis to prolonged
cholestasis, hepatomegaly, and severe liver dysfunction [8]. The
diagnostic criteria of CFLD comprises either a positive
histopathological test (focal or multilobular biliary cirrhosis)
or presence of at least two of the following criteria, evaluated
at least twice a year: (i) Hepatomegaly (>2 cm below the costal
margin in the midclavicular line) confirmed by ultrasound test;
(ii) abnormal elevation of liver enzymes; and (iii) positive
ultrasound findings (increased echogenicity of liver parenchyma,
tuberosities, irregular edges and splenomegaly) [8,10].
Pulmonary complications are the predominant cause of morbidity
and mortality in CF. CFLD is an evolving paradigm and is
believed to the third commonest cause of mortality in patients
with CF [8].
The symptoms evolve over time, and in early
infancy, anemia may be the first clinical presentation of CF
[3]. These infants typically have normocytic normochromic anemia
secondary to multiple etiologies like iron deficiency, chronic
inflammation, vitamin E deficiency, ineffective erythropoiesis,
and ongoing micro-bleeding. The anemia is often accompanied by
hypoalbuminemia [11]. The severity of hypoalbuminemia can be
used as a marker of severity of respiratory morbidities in later
life [12]. These two manifestations can precede respiratory
symptoms for many months [3]. Therefore, the concomitant
presence of severe anemia and hypoalbuminemia in early infancy
should raise the possibility of CF.
To establish the
diagnosis of CF, sweat chloride estimation is the first test to
be done, followed by CFTR genetic analysis, and CFTR physiologic
tests. All individuals diagnosed with CF should have a sweat
test and a CFTR genetic analysis performed [13]. However, in
neonates and early infancy, the sweat chloride test is difficult
to perform due to logistic issues; therefore the reliance is
more towards the genetic analysis (CFTR mutation panel).
F508del is the commonest CFTR gene mutation in the Western
population, up to the tune of 80% of all tested alleles.
However, this mutation is much less commonly observed in Asian
and Indian patients [14,15]. Thus, a limited mutation analysis
may not be able to provide a genetic diagnosis of CF, and we may
need complete CF gene sequencing for the confirmation of the
diagnosis.
In the index case, the antemortem diagnosis
could not be made, but post-mortem histopathology along with
positive mutation is diagnostic of cystic fibrosis.
Contributors: JK: clinical protocol discussant, reviewed the
literature and drafted the manuscript; DC: pathology protocol
discussant, reviewed the literature and edited the manuscript;
SL: treating unit consultant, provided critical inputs in the
draft of the manuscript, and edited the manuscript; PK:
substantial inputs in analysis of the case, critically reviewed
and edited the manuscript. All the authors approved the final
version of the manuscript.
Funding: None; Competing
interests: None stated.
Ethics statement: The authors
certify that the family was informed about the final diagnosis,
and appropriate counselling provided.
Annexure I
Name of
Discussants (In the sequence they appear in the manuscript)
Clinical Discussant : Jogender Kumar
Chairperson : KL Gupta
Pediatric Gastroenterologist 1 : Sadhna B Lal
Pediatric
Pulmonologist : Meenu Singh
Neonatologist : Sourabh Dutta
Pediatric Neurologist 1 : Renu Suthar
Pediatric
Gastroenterologist 2 : G. Vybhav Venkatesh
Pediatric
Neurologist 2 : Arundhati B Mukherjee
Pathologist 1 : Kirti
Gupta
Pathology discussant : Debajyoti Chatterjee
Pathologist 2 : Kim Vaiphei
Pathologist 3 : Uma Nahar
References
1. Bhatia V, Bavdekar A,
Matthai J, Waikar Y, Sibal A. Management of Neonatal
Cholestasis: Consensus Statement of the Pediatric
Gastroenterology Chapter of Indian Academy of Pediatrics. Indian
Pediatr. 2014;51:203-10.
2. Saheki T, Song Y-Z. Citrin
Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean
LJ, Stephens K, et al., editors. GeneReviews®. Seattle (WA):
University of Washington, Seattle; 1993. Available from:
http://www.ncbi.nlm.nih.gov/books/NBK1181/. Accessed March 28,
2019.
3. Sismanlar T, Aslan AT, Köse M, Pekcan S, Ezgü
FS, Budakoðlu IÝ, et al. Early severe anemia as the first sign
of cystic fibrosis. Eur J Pediatr. 2016;175:1157-63.
4.
Kabra SK, Kabra M, Lodha R, Shastri S. Cystic fibrosis in India.
Pediatr Pulmonol. 2007;42:1087-94.
5. Davies JC, Alton
EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255-9.
6.
Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med.
2005;352:1992-2001.
7. Elborn JS. Cystic fibrosis.
Lancet. 2016;388:2519-31.
8. Kobelska-Dubiel N,
Klincewicz B, Cichy W. Liver disease in cystic fibrosis.
Przeglad Gastroenterol. 2014;9:136-41.
9. Diwakar V,
Pearson L, Beath S. Liver disease in children with cystic
fibrosis. Paediatr Respir Rev. 2001;2:340-9.
10.
Herrmann U, Dockter G, Lammert F. Cystic fibrosis-associated
liver disease. Best Pract Res Clin Gastroenterol.
2010;24:585-92.
11. Dolan TF, Rowe DS, Gibson LE. Edema
and hypoproteinemia in infants with cystic fibrosis: The
hypoalbuminemia sometimes seen is presumably secondary to
malabsorption. Clin Pediatr (Phila). 1970;9:295-7.
12.
Abman SH, Reardon MC, Accurso FJ, Hammond KB, Sokol RJ.
Hypoalbuminemia at diagnosis as a marker for severe respiratory
course in infants with cystic fibrosis identified by newborn
screening. J Pediatr. 1985;107: 933-5.
13. Farrell PM,
White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al.
Diagnosis of Cystic Fibrosis: Consensus Guidelines from the
Cystic Fibrosis Foundation. J Pediatr. 2017;181:S4-S15.e1.
14. Sharma H, Jollivet Souchet M, Callebaut I, Prasad R,
Becq F. Function, pharmacological correction and maturation of
new Indian CFTR gene mutations. J Cyst Fibros. 2015;14:34-41.
15. Alibakhshi R, Kianishirazi R, Cassiman J-J, Zamani M,
Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis
patients: Identification of eight novel mutations. J Cyst
Fibros. 2008;7:102-9.
|
|
|
 |
|
