|
|
Brief Reports Indian Pediatrics 2006;43:340-343 |
||||
|
Attenuated Form of Evans Syndrome Among Pediatric ITP Patients |
||||
|
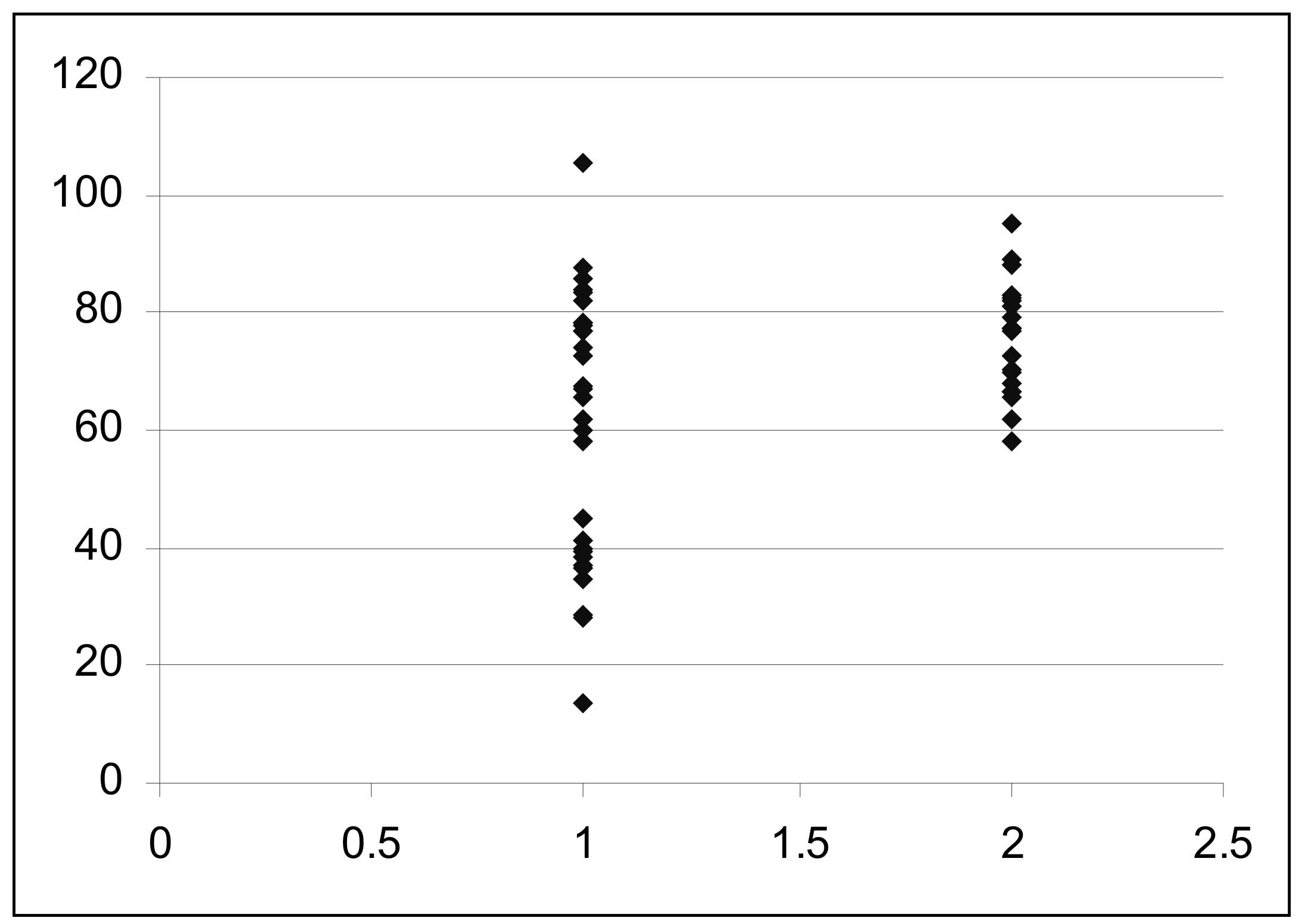
Idiopathic thrombocytopenic purpura (ITP) or primary thrombocytopenic purpura, is diagnosed when no clinically apparent associated conditions or other causes of thrombocytopenia are present. There is no specific criteria to establish the diagnosis of ITP. The low platelet count in ITP is secondary to rapid immune mediated platelet destruction by anti platelet antibodies directed against platelet membrane glycoproteins(1). ITP may be primary or is associated with variety of immunologic disorders where thrombocytopenia is part of a clinically overt or occult autoimmune disease. Patients with antinuclear or antiphospholipid antibodies and thrombocytopenia with no clinical evidence of SLE, are considered to have ITP in both adult and pediatric age groups(2-3). Similarly in Evans syndrome, thrombocytopenia is associated with hemolytic anaemia due to auto antibodies against red cells(4). In this study we assessed evidence of hemolysis by a sensitive method of estimation of free serum haptoglobin, in pediatric ITP patients with no clinical feature(s) of hemolytic anaemia, or positive direct Coombs’ test. Subjects and Methods This study included 30 patients (23 males and 7 females), age 6 months to 17 years, seen between August 2003 till March 2004, newly diagnosed to have ITP. The diagnosis was made from medical history , detailed physical examination, low platelet count (<100,000/cmm), a normocytic normochromic peripheral blood smear and increased mega-karyocytes in the bone marrow examination. Patients referred with bleeding complaints (easy bruisability, echymosis, purpura, mucosal bleeding) and found to have isolated thrombocytopenia with no cause to account for, were included in the study. Microangiopathic hemolytic anemia (MAHA), SLE and overt Evans syndrome were excluded by appropriate laboratory tests. These included prothrombin time, activated thromboplastin time, fibrin degradation products, ANCA, direct Coombs test (DCT), and serum haptoglobin determined by chromatography(5) in the patients, and 20 age matched normal controls. All tests were performed prior to initiating any treatment, and none had prior history of any specific medication including corticosteroid therapy. Results There were 23 males and 7 females in this study. Their age ranged from 6 months to 17 years (median 5 years). Duration of the illness was 1 day to 1.5 years (median 7 days). All patients had either rash/blue spots/petechae or echymosis at presentation. Besides 3 had epistaxis, five had fever and one each had ocular bleed and hematemesis. Their hemoglobin ranged from 8.3 to 14.2 g/dL. (mean 10.5g/dL). Platelet counts ranged 9000-65000/cmm (median 18000/cmm). Bone marrow was normo cellular in 26, mildly hypo cellular in 3 and normoblastic in 23 and mildly megaloblastic in 7. Marrow megakaryocytes were increased in all cases. All patients were negative for ANCA, MAHA, and DCT. Serum haptoglobin in the control subjects ranged from 58.05 to 95.03 mg/dL (mean 75.53 ± 12.67 mg/dL), while in patients of ITP it ranged from 13.39 to 105.52 mg/dL (mean 60.86 ± 22.77 mg/dL). In 19 patients, serum haptoglobin was very similar to that in the controls (range 58.14-105.52 mg/dL, mean 76.00 ± 12.7 mg/dL). However in 11 (36.7%) patients with ITP, serum haptoglobin was low, ranging from 13.39 to 44.91 mg/dL, mean 34.74 ± 8.3 mg/dL, P <0.001, (Fig 1). No statistically significant differences were observed in gender ratio, initial platelet counts, Hb levels, reticulocyte counts, type of erythropoiesis or response to treatment in those with normal or low levels of haptoglobin.
Discussion Idiopathic thrombocytopenic purpura (ITP) is a syndrome of primary acquired thrombocytopenia in the absence of bone marrow failure, in which platelets are destroyed by the hosts’ immune system. It may occur as part of an immune disorder affecting other systems. In Evans syndrome thrombocytopenia is associated with auto-immune hemolytic anemia. A fully developed Evans syndrome should be associated with a Coomb’s positive hemolytic anemia (i.e., antibodies against red cells), along with low platelet count. However, when antibody levels are low, a degree of hemolysis may be present without any overt evidence of hemolytic anemia and a negative direct Coomb’s test, qualifying for a form such as ‘attenuated Evans syndrome’. In the present study 11 of the 30 cases of ITP (36.7%) showed abnormally low levels of free serum haptoglobin, signifying a degree of hemolysis occurring in these cases. Haptoglobin is a glycoprotein which is synthesized in the liver and circulates in blood. It consists of two pairs of a chains and two pairs of b chains. Free hemoglobin as a result of hemolysis, readily dissociates into dimers of a and b chains. The a chains bind avidly with the b chains of haptoglobin in plasma or serum to form a complex to be rapidly removed from circulation and catabolized by the liver parenchymal cells. Haptoglobin begin to be depleted when the daily hemoglobin turnover exceeds about twice the normal. When the plasma hemoglobin exceeds 30 to 200 mg/dL, which is the capacity of haptoglobin to bind hemoglobin, the free ab dimers of hemoglobin readily pass through the glomerulus of the kidney. Decrease in haptoglobin content from the plasma of patients who have accelerated red cell destruction occurring primarily within macrophages may occur due to excess haemoglobin which leaks from phagocytic cells into the plasma to bind to haptoglobin(6). Thus, the measurement of free plasma haptoglobin is useful in assessing the hemolysis before other routinely performed laboratory tests depict clear evidence. Asessing plasma free haptoglobin, Ruiz-Arguelles(7) reported low levels in 5 of their 17 (29%) adult patients with ITP. In their study the long-term thrombocytopenia-free status was lower in patients with low haptoglobin levels. Patients with ITP can show highly variable clinical courses during long-term follow-ups. Some of them develop secondary autoimmune disorders such as Evans syndrome(8). In a long term follow up study of 72 Asian adult patients with ITP(9), thirteen (18%) patients developed secondary auto-immune diseases, which included 3 cases of Evans syndrome. These authors also concluded that secondary autoimmune disease impact adversely on their prognosis. Alter-natively, among pediatric autoimmune hemolytic anemia cases, overall mortality rate was higher in those with associated autoimmune thrombocytopenia i.e., Evans syndrome(10). We expect to clarify some of these issues in the long-term follow up of our cases. Our data in pediatric patients added to the previous evidence in adult patients(7), support the observation of Evans made 50 years ago, "there is a spectrum-like relationship between primary thrombocytopenia and hemolytic anemia"(4). Thus, the concept of attenuated form of Evans syndrome is made out for clinical application. Contributors: SD conceived, planned, executed this study and has wrote the manuscript. RM and AT contributed the patients’ data and material for this study. Funding: PGIMER. Competing interests: None stated.
| ||||
|
References | ||||
|
|
![]()