Mamta Manglani
M.R. Lokeshwar
Ratna Sharma
From the Division of Pediatric Hematology-Oncology,
Department of Pediatrics, L.T.M.M. College and General Hospital, Sion,
Mumbai, India.
Correspondence to: Dr. Mamta Manglani, 51, Sea
Springs, B. J. Road, Band Stand, Bandra (W), Mumbai 400 050, India.
Manuscript received: July 22, 2002; Initial review
completed: September 19, 2002;
Revision accepted: December 23, 2002.
Diamond-Blackfan anemia is a rare congenital
hypoplastic anemia. We report 6 children diagnosed as Diamond-Blackfan
anemia at our clinic. All had severe pallor at presentation, with mild
hepato-megaly and just palpable spleen in one child. Thumb anomaly was
present in one of them. All of them had macrocytic or normocytic
anemia with reticulo-cytopenia, and bone marrow examination revealed
marked erythroid hypoplasia. All of them were treated with oral
steroids with a good response.
Key words: Congenital pure red cell aplasia, Diamond-Blackfan
syndrome, Oral steroids.
Diamond-Blackfan anemia (DBA) is a rare congenital
hypoplastic anemia that usually presents early in infancy. Congenital
anomalies, in particular of the head and upper limbs, are present in
25% of reported patients(1). DBA has also been referred to as
congenital pure red cell aplasia, chronic aregenerative anemia,
erythrogenesis imper-fecta and congenital hypoplastic anemia(2). The
diagnosis of this disease is clinched by the finding of a normochromic,
macrocytic or normocytic anemia manifesting early in life,
reticulocytopenia, normocellular bone marrow with erythroid hypoplasia.
To date, a total of approximately 600 patients have been reported
worldwide. 354 patients have been enrolled in the Diamond Blackfan
Anemia registry of North America and 229 in the DBA group of France,
Germany and eight other countries(3). We report six such cases
diagnosed at our clinic.
Case Report
Six children, four males and two females were seen
with a history of pallor noticed between two and four months of age
(Table I). Four of these patients came with the incorrect diagnosis of
iron deficiency anemia (IDA). They presented to us at the age of
7,8,10 and 18 months. One child was clinically diagnosed as
thalassemia major elsewhere and was seen at our clinic at 7 months of
age, while the 6th child had received a transfusion at the age of two
months for anemia with cardiac failure, without any investigations for
the cause of anemia. This child came to us at the age of 10 months.
All of them had received transfusions ranging from one to four times
before being seen at our clinic. There was no history of consanguinity
amongst parents and none of the patients had affected siblings. The
antenatal and perinatal periods were uneventful in all of them. None
of them were small for gestational age at birth.
Table I
Clinical Features and Investigative Findings in Six Patients with Diamond
Blackfan Anemia.
|
Case 1
|
Case 2
|
Case 3
|
Case 4
|
Case 5
|
Case 6
|
Age at presentation (mo)
|
7
|
8
|
10
|
18
|
7
|
10
|
Age of 1st symptom (mo)
|
2
|
2.5
|
2.5
|
3
|
4
|
2
|
Gender
|
Male
|
Male
|
Male
|
Female
|
Female
|
Male
|
Pallor
|
Present
|
Present
|
Present
|
Present
|
Present
|
Present
|
Congenital anomalies
|
Absent
|
Absent
|
Absent
|
Absent
|
Thumb anomaly
|
Absent
|
Hepatomegaly
|
Yes
|
Yes
|
Yes
|
Yes
|
Yes
|
Yes
|
Splenomegaly
|
No
|
No
|
No
|
JP
|
No
|
No
|
Hemoglobin (g/dL)
|
3.0
|
5.4
|
3.8
|
4.4
|
6.0
|
4.8
|
Platelet count (lacs/c.mm)
|
4.34
|
3.5
|
3.68
|
6.3
|
5.54
|
3.5
|
MCV (fL)
|
91
|
98
|
99
|
88
|
79
|
94
|
Reticulocyte
|
0.5
|
0.6
|
0.3
|
0.5
|
0.8
|
0.4
|
HbF (%)
|
2.5
|
2.2
|
5.8
|
6.8
|
0.5
|
8.6
|
Transferrin saturation (%)
|
22
|
38
|
33
|
45
|
53
|
40
|
M:E ratio in bone marrow
|
16:1
|
16.5:1
|
20:1
|
19.5:1
|
20:1
|
18:1
|
JP: Just Palpable.
On examination, all had severe pallor and mild
hepatomegaly. Spleen was just palpable in one patient. One of the
patients had thumb anomaly. No other congenital anomalies were
observed during physical examination in any of them. Other systems
were normal. Investigations revealed hemoglobin ranging from 3 to 6 g/dL,
with a corrected reticulocyte count of less than 1%. The peripheral
smear showed macrocytic (4 patients) or normocytic (2 patients) and
normochromic anemia with mild anisocytosis. The MCV at diagnosis
ranged from 79 to 99 fL. Leucocyte counts were normal in all patients,
however, platelet counts were high in 2 patients. All patients had
elevated serum iron levels with transferrin saturation ranging between
22 and 53%. Fetal hemoglobin (HbF) was elevated in 5 of them ranging
from 2.2% to 8.6%. Only one child had HbF of 0.5%. Skeletal surveys
and ultrasonography of the abdomen ruled out any congenital anomalies,
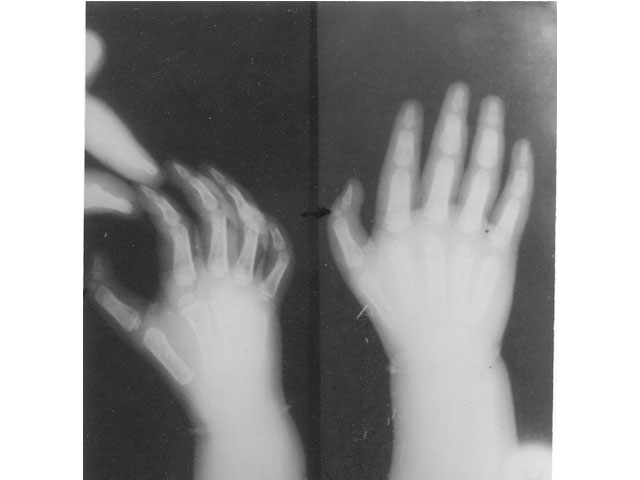
except in the one child (case 5) with thumb anomaly. Her X-ray showed
an accessory bone in the left thumb with a normal biphalangeal
configuration of the right thumb (Fig. 1). Chromosomal studies were
normal in the 5 patients, in whom it could be done.
 |
|
Fig. 1. X-ray of hands (case 5) showing an
accessory bone between the 2 phalangeal bones in the left thumb and
normal biphalangeal configuration of the right thumb
|
The diagnosis was confirmed on bone marrow
aspiration and trephine biopsy, which showed marked erythroid
hypoplasia with elevated M:E ratio ranging from 16:1 to 20:1.
Megakaryopoiesis and myelopoiesis were normal in all. All of them
required one transfusion of packed red cells of 5 mL/kg body weight,
following which they were given oral prednisolone in the dose of 2
mg/kg body weight/day in three to four divided doses. A good response
was noted in all 6 patients with a rise in hemoglobin (6 to 9 g/dL)
and reticulocyte counts (1 to 3%) within a preriod of 7 to 10 days of
therapy. After the hemoglobin rose to 10 g/dL, they were shifted to
alternate day low dose therapy with tapering of the doses. Three
patients have been maintaining hemoglobin between 11 and 12.5 g/dL on
intermittent low dose alternate day steroid therapy for a median
follow-up of 4.3 years (range: 4 to 8 years). Two have sustained
remission and are off steroid therapy, after initial therapy for 4 and
8 months each (follow up of 2.3 and 4 years). One patient has been
lost to follow up.
Discussion
Congenital pure red cell aplasia was first
described by Josephs in 1936 and two years later in 1938, Diamond and
Blackfan reported four such cases(2). DBA is usually seen in infancy,
although cases have been detected as late as at 6 years of age. The
male to female ratio 1.2:1.
Approximately 1/4th to 1/3rd of patients have
congenital anomalies involving the upper limbs and the head, and the
urogenital or cardiovascular systems. However, the link between these
malformations and defective erythropoiesis is unclear and a defect in
a molecule acting on both early embryonic development and
hematopoiesis has been proposed(4). Most cases are sporadic with
inheritance observed in about 10% of patients, with a dominant or,
more rarely, recessive pattern. One locus on chromosome 19q13.2
encoding ribosomal protein S19 accounts for a quarter of patients with
either the dominant or the sporadic form(1,4). Another locus in
association with DBA has been detected on chromosome 8p(5). Families
not linked with either of these loci have also been described(4,5).
The basic defect is hypothesized to be a decrease
in number or function of erythroid precursors, both the CFU-E and
BFU-E(4). A high level of erythropoietin in the serum suggests
progenitor insensitivity to erythro-poietin(6). Additionally,
immunologic cellular factors have also been considered responsible for
the pathogenesis in DBA(7). However, anecdotal studies refute this
finding(8).
The closest differential diagnosis of this syndrome
is TEC (transient erythro-blastopenia of childhood). However, the mean
age of diagnosis of TEC is 26 and 28 months in males and females
respectively, whereas that for DBA is 5.2 and 6.6 months(2). MCV is
generally normal in TEC and increased in DBA, and a history of
preceding viral illness with no congenital anomalies characterize
TEC(2). HbF is generally normal at diagnosis in TEC, whereas it is
high in DBA. Besides, TEC recovers spontaneously within 1 to 2 months
of onset, irrespective of treatment(2).
Red cell transfusions were the only available
modality of treatment in the earlier times. Though, it still remains
an important form of therapy for those who are refractory to other
medications, several newer phar-macologic agents have been found
effica-cious. Apart from the conventional oral prednisone (overall
response rates of 60% to 70%) and high-dose intravenous methyl-prednisolone,
various drugs have been tried with limited success(2). These include
androgens, danazol, 6-mercaptopurine, cyclo-phosphamide,
antilymphocyte globulin, vincristine etc.(2). Cyclosporine has been
tried with conflicting results in different studies(9). Allogeneic
stem cell transplanta-tion with HLA-identical related donor has shown
promising results, though anecdotal reports of failure have been
reported(10). Prenatal diagnosis has been constrained by the fact that
DBA is inherited only in 10% of cases.
Acknowledgement
The authors wish to thank Dr. M.E. Yeolekar, Dean,
LTMG Hospital and College, Sion, Mumbai for permitting the publication
of this manuscript. We also wish to acknowledge the support received
from our Head of the Department, Dr. Madhuri Kulkarni during the
preparation of this manuscript.
Contributors: MM diagnosed and managed these
patients. She prepared the manuscript and finalized the draft. MRL
guided in patient care and revised the draft. RS helped in diagnosis
and management of these children and assisted in modifying the draft.
Funding: None.
Competing interests: None stated.
1. Willig TN, Gazda H, Sieff CA. Diamond-Blackfan
anemia. Curr Opin Hematol 2000; 7: 85-94.
2. Alter BP, Young NS. The Bone Marrow Failure
Syndromes. In: Nathan and Oski’s Hematology of Infancy and
Childhood. Eds. Nathan DG, Orkin SH, 5th edn. 1998; pp 238-335.
3. Vlachos A, Klein GW, Lipton JM. The Diamond
Blackfan Anemia Registry: tool for investigating the epidemiology and
biology of Diamond Blackfan anemia. J Pediatr Hematol Oncol 2001; 23:
377-382.
4. Dianzani I, Garelli E, Ramenghi U. Diamond-Blackfan
Anemia: an overview. Pediatr Drugs 2000; 2: 345-355.
5. Gazda H, Lipton JM, Willig TN, Ball S, Niemeyer
CM, Tchernia G et al. Evidence for linkage of familial Diamond-Blackfan
anemia to chromosome 8p23.3-p22 and for non-19q non-8p disease. Blood
2001; 97: 2145-2150.
6. Tsai PH, Arkin S, Lipton JM. An intrinsic
progenitor defect in Diamond-Blackfan anemia. Br J Haematol 1989; 73:
112-120.
7. Sawada K, Koyanagawa Y, Sakurama S. Nakagawa S,
Konno T. Diamond-Blackfan syndrome: a possible role of cellular
factors for erythropoietic suppression. Scand J Hematol 1985; 35:
158-165.
8. Freedman MH, Saunders EF. Diamond-Blackfan
syndrome: evidence against cell-mediated erythropoietic suppression.
Blood 1978; 51: 1125-1128.
9. Alesandri AJ, Rogers PC, Wadsworth LD, Davis JH.
Diamond-Blackfan anemia and cyclosporine therapy revisited. J Pediatr
Hematol Oncol 2000; 22: 176-179.
10. Vlachos A, Federman N, Reyes-Haley C, Abramson J, Lipton JM.
Hematopoietic stem cell transplantation for Diamond-Blackfan anemia: a
report from the Diamond Blackfan Anemia Registry. Bone Marrow
Transplant 2001; 27: 381-386.
|
