|
|
Case Reports Indian Pediatrics 2001; 38: 1045-1049 |
||
|
Joubert Syndrome |
||
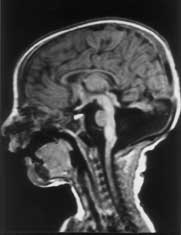
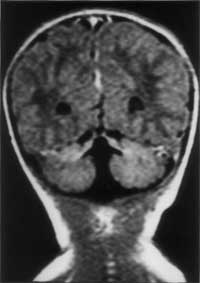
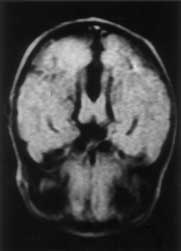
Joubert syndrome is a rare autosomal recessive disorder characterized by abnormal respiratory pattern and eye movements, hypotonia, ataxia, developmental retardation with neuropathologic abnormalities of cerebellum and brainstem. We report a case of Joubert syndrome presenting in the neonatal period. Although previously described in Indian literature(1), this is probably the first case report of Joubert syndrome in a neonate from our country. Case Report A 12-hour-old term baby boy born to a second gravida mother was admitted with history of respiratory distress since 4 hours of age. The infant was the product of a second degree consanguineous marriage. The first pregnancy of the mother ended in an intrauterine death at 8 months of gestation. The baby was delivered normally after an uneventful pregnancy and cried immediately after birth. The birth weight was 2430 g and head circumference 32.0 cm. On admission the baby was found to have episodes of panting type of breathing with respiratory rate upto 160 breaths/minute, alternating with apnea lasting for 10-15 seconds, without cyanosis or alteration in heart rate. He was hypotonic and had intermittent jerky horizontal as well as vertical eye move-ments. The pupils were equal and reacting to light. General physical examination did not reveal any dysmorphism and systemic examination was normal. The following investigations were carried out and found to be normal: Complete hemo-gram, blood urea, blood sugar, serum calcium, electrolytes, lactate, ammonia, chromosomal studies, plasma and urine amino-acids. Blood culture was sterile. X-ray chest and examina-tion of fundus were normal. Echocardiography and ultrasonography of abdomen were normal. Arterial blood gas estimation revealed uncompensated respiratory alkalosis. Computed tomogram (CT) of brain showed sagittal vermic cleft communicating with fourth ventricle and cisterna magna, consistent with aplasia of vermis. Magnetic Resonance Imaging (MRI) revealed complete aplasia of vermis (Figs. 1 & 2) with deep interpeduncular fossa and thick-ened superior cerebellar peduncles producing a "molar tooth appearance" (Fig. 3). The infant was followed up in the High Risk Infant Clinic of our department at 3 months of age. He was being exclusively breast-fed and velocity of growth was within normal limits. At this time, though social smile was attained he was still hypotonic and had no head control. The abnormal ocular movements and periodic hyperventilation were seen only occasionally when the child was awake and disappeared during sleep.
Discussion Joubert syndrome is a rare and probably under-diagnosed syndrome with a bad prog-nosis. Marie Joubert and associates in 1969 were the first to describe this syndrome which included episodic hyperpnea, abnormal eye movements, ataxia and mental retardation with agenesis of cerebellar vermis in 4 siblings and 1 sporadic case(2). Since Joubert’s initial report, more than 100 patients have been described in the literature. Inheritance of this disease is said to be autosomal recessive. Recent studies have concluded that it is a genetically heterogenous disorder with one locus mapping to chromosome 9q(3,4). In Joubert syndrome, midline structures of the brain-stem have both structural and func-tional defects(5). Neuropathological studies reveal agenesis of cerebellar vermis, hypo-plasia or fragmentation of several brainstem nuclei and dysplasia of structures at the ponto-mesencephalic junction. Extensive brainstem malformation could explain the oculomotor apraxia and hyperpnea; anomalies of the gracile nuclei and solitary tract are thought to contribute to the abnormal respiratory pattern(6). The breathing pattern in Joubert syndrome is effortless hyperventilation (upto 200 breaths/min) which is more conspicuous in awake state and intensifies when the patient is stimulated, interspersed with central apnea. This abnormal breathing pattern usually wanes with age. The eye abnormalities commonly observed in this disease are complete oculomotor apraxia, both in horizontal and vertical direc-tions and ocular coloboma. Severe visual loss, pendular nystagmus, pigmentary changes in fundus and decreased vestibulo-ocular reflexes are seen in a subset of individuals and are thought to be a form of Leber’s amaurosis(7). Hypotonia and mental retardation are constant features of Joubert syndrome. Long-term followup of the children with this disease reveal that majority had severe neurodevelop-mental impairments(8). Common malforma-tions associated with Joubert syndrome are polydactyly, renal cysts, and soft tissue tumors of the tongue(3,9). The hallmark imaging features of Joubert syndrome are: (i) Dysgenesis of the isthmus (part of the brainstem between pons and inferior colliculus) which is seen as elongation and thinning of ponto mesencephalic junction, and deep interpeduncular fossa; (ii) thickening of superior cerebellar peduncles; (iii) hypo- plasia of vermis characterized by incomplete lobulation and enlarged fourth ventricles; (iv) incomplete fusion of the halves of the vermis creating a sagittal vermis cleft seen on axial or coronal MRI planes. Combination of the first 3 features produce the characteristic "molar tooth sign" on Axial MRI imaging(10). Evaluation of a child with suspected Joubert syndrome should include MRI scan, retinal examination, renal ultrasound, electro-retinogram and karyotyping(11). It should be noted that CT or MRI finding of vermis hypo-plasia in the absence of other typical clinical features even with mental retardation does not lead to the diagnosis of Joubert syndrome. Prognosis of this disease is usually poor with hypotonia and severe developmental delay. Contributors: RS designed the paper and prepared the manuscript. AKJ was the overall co-ordinator and guide; he will act as the guarantor for this paper. SS helped in radio imaging. AB helped in compiling the data and drafting the paper.
Funding: None
| ||
| References | ||
|
![]()