|
|
Original Articles Indian Pediatrics 2000;37: 939-946 |
||
|
Krabbe Disease - Clinical Profile |
||
|
Manuscript received: December 28,
1999; Initial review completed: February 8, 2000;
Leukodystrophies are a group of rare inherited neurometabolic disorders resulting from defects in the synthesis or catabolism of myelin. Those with an early infantile onset include – Krabbe disease (KD) or globoid cell leukodystrophy (GLD), Pelizaeus-Merzbacher disease, Canavan disease, and Alexander disease. KD is a rare autosomal recessive disorder resulting from the deficiency of the lysosomal enzyme, galactocerebrosidase (GALC) which catabolises degradation of several galactolipids such as galactosylceramide, galactosylsphingosine and galactosyldi-glyceride(1). Early as well as late infantile forms of the disease are well known, but juvenile and adult onset disease are now being increasingly reported. Clinical and radiological manifestations in these age groups are hetero-geneous and the clinician should be aware of these differences. There is no data on KD from India following a MEDLINE search from 1985 to 1999. We present a series of 9 cases of enzy-matically confirmed KD with emphasis on the clinical spectrum, neuroimaging and enzyme diagnosis.
Hospital records of children diagnosed to have KD over a period of 2 years at the Genetic Clinic of a tertiary care teaching hospital in Mumbai were reviewed. Demographic data, clinical features and results of investigations (including neuroimaging) were recorded. GALC activity in peripheral blood leukocytes was assessed photometrically(2) and the values were compared with those obtained from age matched controls. All the samples and controls were tested for the enzyme Aryl sulfatase-A to ensure quality control. TNPAL-galactocerebro-side (115 nmol), 0.5 mg crude sodium tauro-cholate and 60 mg oleic acid in 25 mmol citrate buffer at pH 4.5 was used as substrate.
Nine children (7 females, 2 males) identified on the basis of clinical features and investigations as possible KD, had low levels of GALC in their leukocytes. Based on the age of onset of the disease and clinical presentation, 5 patients had the classical infantile form of the disease, 3 belonged to the late infantile group and a single case who presented at 8 years of age was diagnosed to have the juvenile form of KD. Infantile KD Of the total 8 cases (5 classical and 3 late infantile types), 6 were females and 2 were males. Mean age at presentation was 9.4 months with a range of 2.5 to 21 months. The clinical features are summarized in Table I. Consanguinity was absent in all the cases. Cases 2, 4 and 5 had affected (though undiagnosed) siblings. Diag-nostic protocol in these cases included cerebro-spinal fluid (CSF) analysis, nerve conduction (NC) studies, neuroimaging (CT scan and/or MRI) and GALC assay in peripheral blood leukocytes. Table I gives the results of these investigations. EEG was done in 3 cases (Case 1, 6 and 8) and was abnormal in Case I (showing left temporo-parietal epi-leptiform transients) and Case 8 (showing left sided paroxysmal activity and subcortical epileptogenic activity with solitary right burst). Additional investigations done in isolated cases included brainstem evoked response audiometry (BERA) (Case 1) and visual evoked potentials (VEP) (Case 5) which were normal. None of the cases were available for follow up except Case 1 who was admitted for bronchopneumonia and subse-quently died at 7½ months of age. TABLE 1–Clinical
Features and Investigations in Infantile Krabbe Disease. |
|
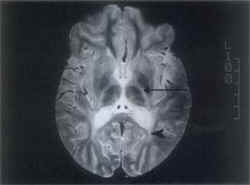
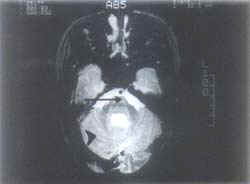
ND = Neurodegeneration; MS = Milestones; %ile = Percentile. Juvenile KD An 8-year-old female was born of a non-consanguineous marriage with normal antenatal and birth history. She had delayed mental and motor milestones since the age of 18 months. On general examination, her weight was 20.5 kg (5th-10th percentile), height was 112 cm (<5th percentile) and head circumference was 48.5 cm. The developmental quotient was 11 months. Hypertelorism, flat feet and knee joint flexion contractures were noted. Central nervous system examination revealed spastic quadriparesis with depressed deep tendon reflexes. Plantars were flexor in response and there were no cerebellar signs. Relevant investigations were as follows: Leukocyte GALC level was undetectable, MRI (brain) showed white matter involvement (Figs. 1 & 2), NC studies revealed a diffuse demyelinat-ing sensorimotor neuropathy, audiometry showed bilateral sensorineural hearing loss and VEP was normal.
Globoid cell leukodystrophy (GLD) or Krabbe disease (KD) owes its name to the globoid cells (which are characteristic storage cells) found infiltrating around cerebral blood vessels. In his report in 1916, Krabbe described this disease as ‘diffuse cerebral sclerosis’(3). Though the metabolic nature of the disorder was recognized in the late fifties, the etiological basis was established later by Suzuki and Suzuki in 1970 following the demonstration of GALC deficiency in the tissues of affected patients(4). The gene has been cloned and localized to the q21-q31 region of chromosome 14(1). The molecular basis of the disease has been recently established by the identification of a number of mutations and an attempt is being made to establish a genotype-phenotype correlation(5). A simple classification takes into account the age at onset of the disease and reflects the variations in clinical presentations of the subtypes(6). The classical infantile form mani- fests in the first six months of life with neurodegeneration, failure to thrive, fever, seizures and visual loss. Initial hypotonia gives way to spasticity. As a rule, death occurs by 2 years of age. As opposed to this, a more protracted course with onset of gait disturbances in the second year of life, progressive spasticity and visual loss are characteristics of the late infantile form of the disease. Pyramidal weakness and blindness predominate in the juvenile form, whereas adult onset disease is characterized by slowly progressive ataxia or spasticity with preservation of intellect(7). Hypertelorism though not a feature of KD, was seen in 6 out of 9 cases in our study. Investigations too reflect the dissimilarities between the various subtypes– CSF analysis with raised proteins and NC studies demonstrating peripheral neuropathy are the hallmarks of infantile and sometimes juvenile disease, but adult onset disease is rarely if ever characterized by peripheral neuropathy and raised CSF proteins(6-8). In the absence of confirmatory evidence of low or absent GALC levels, the characteristic distribution of lesions and MRI signal patterns on T2-weighted images can be diagnostic. Invove-ment of pyramidal tracts, parieto-occipital white matter, posterior corpus callosum, and cerebellar white matter involvement as well as lesions of deep grey matter and cerebral atrophy may be seen(9). Prior to availability of an enzyme assay, definitive diagnosis was based on histopatho-logical demonstration of globoid cells by brain biopsy. This pathognomonic PAS-positive cell of monocytic origin contains tubular crystalloid aggregates on electron microscopy(1). The demonstration of needle-like inclusions and splinter shaped structures in eccrine sweat glands of axillary skin specimens by electron micro-scopy provides a relatively non-invasive and safe alternative to brain or nerve biopsy(10,11). GALC assay for the diagnosis is expensive and not widely available. Enzyme estimation in cultured skin fibroblasts or peripheral blood leukocytes accurately identifies homozygotes, but its value in heterozygote detection is limited(12). Mutation analysis could prove to be vital in identifying the hetero-zygotes(13,14). Therapy for KD in the early stages with hematopoietic stem cell transplantation has been successfully attempted with radiological and clinical improvement(15). Retrovirus-mediated gene transfer is found to correct the enzyme deficiency experimentally and holds promise as futuristic therapy(16). At present, counseling and prenatal diagnosis (by enzyme estimation from cultured amniocytes, skin fibroblasts or fetal tissues like brain, kidney, and liver) are the only management strategies that can be offered(17). Of note, has been the successful engraftment after in-utero bone marrow transplant in an affected fetus(18). In conclusion, the specturm of KD varies from a rapidly progressive disease in infancy with a fatal outcome to a more benign course with gait disturbances in adolescents and adults. In our series, children selected on basis of suggestive clinical symptoms demonstrated the characteristic features of KD on neuroimaging; all proved to have undetectable or low levels of GALC in peripheral blood leukocytes. Diag-nosis by enzyme estimation is not yet available at most of the centers specializing in neuro-metabolic disease in India. In this situation, neuroradiologic features on MRI can be relied upon to establish the diagnosis.
The authors thank Dr. R.G. Shirahatti, Dean, Seth G.S. Medical College and KEM Hospital, Mumbai, for granting permission to publish this article. The authors also thank Dr. Meher Ursekar, Consulting Radiologist, Bombay Hospital, Mumbai, for her help in reporting of the MRI scans. Contributors: MST participated in data collection, analysis and drafting the paper. MNM participated in data collection and drafting the paper and will act as guarantor for the paper. PPK participated in the enzyme analysis of all the cases and helped in drafting the paper. BAB was instrumental in the diagnosis and investigations of the cases reported. Funding:
None.
|
![]()