|
|
|
Indian Pediatr 2015;52: 805 -806 |
 |
Novel Mutations in a Patient with Triple A
Syndrome
|
|
Jyoti Sanghvi, Ajit Anand Asati, *Ravindra Kumar and
#Angela Huebner
From Department of Pediatrics and *Central Research
Laboratory, Sri Aurobindo Medical College and PG Institute, Indore,
Madhya Pradesh, India; and #Klinik für Kinder- und Jugendmedizin,
Technische Universität Dresden, Germany.
Correspondence to: Dr Jyoti Sanghvi, Department of
Pediatrics, Sri Aurobindo Medical College and PG Institute, Indore,
Madhya Pradesh, India.
Email: [email protected]
Received: January 09, 2015;
Initial review: March 02, 2015;
Accepted: July 15, 2015.
|
|
Background :
Triple A syndrome (Allgrove syndrome), a rare autosomal recessive
disorder, is characterized by adrenal insufficiency, achalasia cardia
and alacrimia. It is caused by mutations in AAAS gene which
encodes a protein called ALADIN. Case characteristics: 8-year-old
boy who presented with hypoglycemic seizures, dysphagia, dry eyes and
hyperpigmentation. Investigations confirmed achalasia cardia and adrenal
insufficiency. Sequencing of AAAS gene revealed two novel
mutations in compound heterozygous state (c.1101delG/ c.1310_1311delCT).
Outcome: Patient was managed with hydrocortisone and artificial
tears. Message: Sequencing analysis should be done to confirm the
diagnosis of clinically suspected Triple A syndrome.
Keywords: Achalasia cardia, Adrenal
insufficiency, Alacrimia, Allgrove syndrome.
|
|
T
riple A syndrome is characterised by the triad of
adrenal insufficiency, achalasia and alacrimia [1]. In addition a
variety of neurological problems affecting the central, peripheral and
autonomic nervous system may be present [2]. Triple A syndrome is caused
by the mutation in the AAAS gene which encodes for the protein
ALADIN, a constituent of the nuclear pore complex whose function is not
well understood [3,4]. We report a case of triple A syndrome with two
novel mutations present in a compound heterozygous state.
Case report
An 8-year-old boy born to non-consanguineous parents
presented to us with generalized tonic clonic seizure that lasted for
more than 20 minutes. There was a history of repeated vomiting and
progressive dysphagia – more to fluids than to solids – since two years
of age. In addition, his parents noted no tear formation while crying
since age of two years.
His height (116 cm) and weight (16 kg) were below 3rd
centile, and head circumference was 51 cm (between 3 rd
and 50th centile) with a
body mass index of 11.9 kg/m2.
Blood pressure (66/40 mm Hg) was below 5th centile. There was
palmoplantar hyperkeratosis with generalized hyperpigmented skin,
knuckles and gums. Neurological examination revealed nasal speech and
mild intellectual disability. His gait was clumsy and shuffling; pes
cavus was present due to peripheral neuropathy. He also complained of
photophobia.
Laboratory examination revealed hypoglycemia (blood
glucose 25 mg/100mL) Na + 138
mEq/L, K+ 4.3 mEq/L, plasma
ACTH 1863 pg/mL and basal cortisol levels <0.20 µg/dL. Decreased tear
production was recorded by Schirmer test. Barium swallow test showed a
dilation of the esophagus with narrowing of its lower end and tertiary
contracture. Magnetic resonance imaging (MRI) abdomen showed normal
adrenal glands.
A diagnosis of triple A syndrome was made, and for
confirmation the coding sequences of the AAAS gene including
exon-intron boundaries were amplified from genomic DNA and sequenced. We
identified a compound heterozygous AAAS mutation consisting of a
deletion of G in exon 12 at nucleotide position 1101 resulting in a
frame shift at amino acid cysteine 368 as the first affected amino acid,
and a premature stop codon at position 48 of the new reading frame
(c.1101delG; p.Cys368Alafs*48 or short description p.Cys368fs). On the
other allele, we identified a two base pair deletion in exon 14 at
nucleotide position 1310-1311 resulting in a frameshift at amino acid
proline 437 as the first affected amino acid and a premature stop codon
at position 3 of the new reading frame (c.1310_1311delCT;
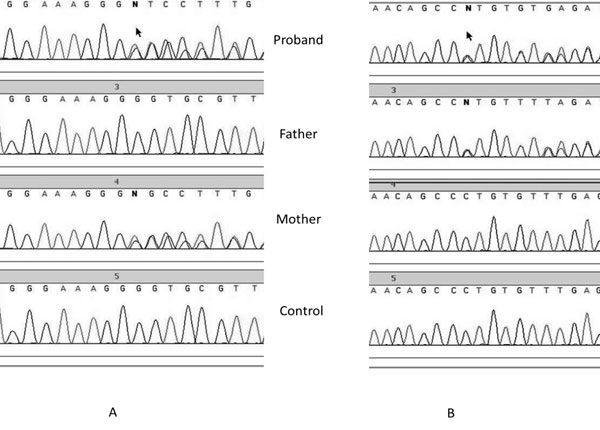
p.Pro437Argfs*3 or short description p.Pro437fs) (Fig. 1).
The mother carried the p.Cys368fs mutation in heterozygous state,
whereas the father was heterozygous for the p.Pro437fs mutation.
 |
|
Fig. 1 Sequence chromatogram of
proband, father and mother showing c.1101delG mutation in exon
12 of AAAS gene in proband and mother(A) and c.1310_1311delCT
mutation in exon 14 of AAAS gene in proband and father.
|
The patient was treated with replacement dose of oral
hydrocortisone (15 mg/m 2/day)
and topical eye lubricants. On regular follow-up after 20 months, the
patient improved and pigmentation decreased significantly.
Discussion
In triple A syndrome, alacrimia is usually the first
manifestation [5]. Achalasia appears with advancing age in two-thirds of
the patients [6]. Adrenal insufficiency normally arises later in life
developing gradually over the first decade, but in some cases
hypoglycemia and seizures may also occur as presenting symptoms
contributing to the diagnosis of the disease. In our case, achalasia
appeared at the age of 2 years and adrenal insufficiency, hypoglycemia
and seizures appeared at the age of 8 years.
Triple A syndrome is caused by mutation(s) in AAAS
gene, located on chromosome 12q13 [7]. Around 68 mutations have been
reported in AAAS gene and most of them produce a truncated
protein, although missense and point-mutations have also been described
[8]. AAAS encodes a protein called ALADIN for alacrimia,
achalasia, adrenal insuficiency and neurological disorder [9]. In the
present case, two novel mutations were identified in a compound
heterozygous state (c.1101delG/ c.1310_1311delCT).
The gene product named ALADIN consists of 546 amino
acids, and belongs to the WD-repeat protein families [3]. The function
of this protein is not clear yet, it is a protein of the nuclear pore
complex (NPC). NPC is critical for communication between the nucleus and
the cytoplasm of cells [10]. However, electron microscopic analysis of
cells from triple A syndrome patients showed no morphologic
abnormalities in NPC, suggesting that mutation in AAAS results in
a functional rather than a structural abnormality in NPC. No specific
genotype-phenotype correlation is found among Triple A syndrome
patients.
Alacrimia is diagnosed by Schirmer test while
achalasia of the cardia and adrenal insufficiency are best diagnosed by
esophageal manometry and ACTH- stimulated cortisol levels, respectively.
Alacrimia is treated with artificial tears while achalasia can be
treated with either pneumatic dilatation or Heller’s myotomy. Adrenal
insufficiency is treated with glucocorticoid and if necessary
mineralocorticoid replacement. However, currently there is no effective
therapy for neurological manifestations.
Contributors: JS: conceived and designed the
study and revised the manuscript for important intellectual content; AA,
RK: collected the clinical data and drafted the manuscript; AH:
performed the molecular analysis.
Funding; None; Competing interests: None
stated.
References
1. Allgrove J, Clayden GS, Grant DB, Macauley JC.
Familial glucocorticoid deficiency with achalasia of the cardia and
deficient tear production. Lancet. 1978;1:1284-6.
2. Clark AJL, Weber A. Adrenocorticotropin
insensitivity syndromes. Endocr Rev. 1998;19:828-43.
3. Tullio-Pelet A, Salomon R, Hadj-Rabia S, Mugnier
C, de Laet MH, Chaouachi B, et al. Mutant WD-repeat protein in
triple-A syndrome. Nat Genet. 2000;26:332-5.
4. Handschug K, Spering S, Yoon SJK, Hennig S, Clark
AJL, Huebner A. Triple A syndrome is caused by mutations in AAAS, a new
WD-repeat protein gene. Hum Mol Genet. 2001;10:283-90.
5. El-Rayyes K, Hegab S, Besisso MA. Syndrome of
alacrima, achalasia, and neurologic anomalies without adrenocortical
insufficiency. J Pediatr Ophthalmol Strabismus. 1991; 28:35-7.
6. Prpic I, Huebner A, Persic M, Handschug K,
Pavletic M. Triple A syndrome: genotype-phenotype assessment. Clin
Genet. 2003;63:415-7.
7. Weber A, Wienker TF, Jung M, Easton D, Dean HJ, Heinrichs
C, et al. Linkage of the gene for the triple A syndrome to
chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet.
1996;5:2061-6.
8. The Human Gene Mutation Database. Available from:
www.hgmd.cf.ac.uk/ac/gene.php?gene=AAAS. Accessed September 30,
2014.
9. Mukhyopadhya A, Danda S, Huebner A, Chacko A.
Mutations of the AAAS gene in an Indian family with Allgrove’s
syndrome. World J Gastroenterol. 2006; 12:4764-6.
10. Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT,
Matunis MJ. Proteomic analysis of the mammalian nuclear pore complex. J
Cell Biol. 2002;158:915-27.
|
|
|
 |
|
