|
|
|
Indian Pediatr 2015;52:
803-804 |
 |
Multiple Pituitary Hormone Deficiency, Empty
Sella and Ectopic Neurohypophysis in Turner Syndrome
|
|
Deep Dutta, Chitra Selvan and Satinath Mukhopadhyay
From Department of Endocrinology and Metabolism,
Institute of Post Graduate Medical Education and Research (IPGMER) and
Seth Sukhlal Karnani Memorial (SSKM) Hospital, India.
Correspondence to: Dr Deep Dutta,
Assistant Professor, Department of Endocrinology, PGIMER and Dr. Ram
Manohar Lohia (RML) Hospital, 1 Baba Kharak Singh Marg, New Delhi
110001, India.
Email: [email protected]
Received: January 07, 2015;
Initial review: March 02, 2015;
Accepted: July 15, 2015.
|
|
Background: Multiple pituitary
hormone deficiency and Turner syndrome have overlapping features in
peripubertal girls and is a diagnostic challenge. Case
characteristics: 16-year-old girl having Turner phenotype undergoing
evaluation for severe short stature and pubertal arrest. Observation:
45,X karyotype, and multiple pituitary hormone deficiency with empty
sella. Intervention: Levothyroxine, growth hormone and
ethinyl-estradiol replacement resulted in 11 cm height gain with
attainment of puberty over 2 years Message: Patients of Turner
syndrome with height <3rd percentile (Turner specific charts) warrant
additional pathology evaluation.
Keywords: Gonadotropin deficiency, Growth
hormone deficiency, Hypothyroidism.
|
|
S
hort stature is a consistent feature in Turner
syndrome [1], and is seen in nearly 100% patients. Ectopic
neurohypophysis and/or empty sella has been observed in 43% children
with growth hormone (GH) deficiency, and is even more common in multiple
pituitary hormone deficiency (MPHD) [2]. MPHD and Turner syndrome have
overlapping features in girls during peripubertal age with both
presenting with short stature and pubertal arrest. We present a child
with MPHD in Turner syndrome.
Case Report
A 16-year-old girl presented with short stature
noticed since 10 years of age along with lack of puberty. She had normal
childhood, without any history of failure to thrive, head injury or
polyuria. Maternal age of menarche was 14 years. Examination revealed
significant short stature (Height 127.2cm; standard deviation. -6.06),
multiple facial nevi (Fig. 1a), cubitus valgus and goiter.
Sexual maturity rating was pre-pubertal. Bone age (Greulich Pyle) was 10
years. Ultrasonography revealed infantile uterus with lack of
visualization of ovaries. Karyotype was 45,X. Height plotted on Turner
syndrome specific growth chart (TSGC) was <5 thpercentile
[1]. Hormonal evaluation revealed secondary hypo-thyroidism and
hypogonadism (low basal and post GnRH analogue stimulated LH). Serum
electrolytes were normal. Anti thyroid peroxidase antibody titer was
elevated (224 U/mL; normal <35 U/mL). Serum IgA anti-tissue
transglutaminase antibody levels were normal (0.2 AU/mL; normal <8 AU/mL).
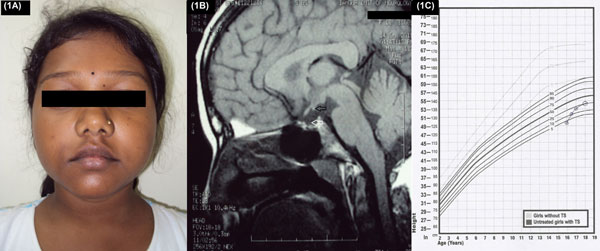 |
|
Fig. 1 (a) Facial profile of
patient showing multiple facial nevi, depressed nasal bridge
with small mandible; (b): MRI brain sagittal section
showing empty sella (white arrow) with ectopic neurohypophysis
at tuber cinerium (black arrow); (c): Height curve of our
patient following levothyroxine, growth hormone and sex steroid
replacement plotted on Turner syndrome specific growth chart.
|
Levothyroxine was started at 50 µg/day. Evaluation of
GH status 4 weeks later revealed GH deficiency
(Web Table I).
Magnetic resonance imaging (MRI) of brain revealed empty sella with
ectopic neurohypophysis near tuber cinerium (Fig. 1b). GH
was started at 3 U/night subcutaneously, increased to 4.5U/night after
three months. Ethinyl-estradiol 2.5 µg/day was also started, along with
calcium and vitamin-D. Reassessment was done 6 monthly.
Ethinyl-estradiol was increased by 2.5 µg every 6 months. She gained 5
cm height in first 6 months, 3 cm in next 6 months, and 3 cm in next 1
year when GH was stopped. Two years after diagnosis, her height was
138.2 cm (Fig. 1c), had B3 breast development, and was on
10 µg of ethinyl-estradiol. Following breakthrough bleeding, patient has
been receiving monthly medroxypro-gesterone along with ethinyl-estradiol
for the last three months, ensuring regular menses.
Discussion
Growth failure in Turner syndrome is characterized by
low IGF-1, increased IGF binding protein-3 proteolytic activity, without
GH deficiency [3]. This report highlights the occurrence of GH
deficiency and MPHD in Turner syndrome. Valenta, et al. [4]
reported two patients of Turner syndrome with hypogonadotrophic
hypogona-dism. There are two other reports of this association [5,6].
Association of MPHD and Turner Syndrome, as in our patients, is even
rarer [7].
Height of all girls with Turner syndrome should be
plotted on TSGC. Those having height <3rd
percentile should be evaluated for additional pathology. Other warning
features include severe short stature, marked bone age delay, and lack
of elevated FSH, as seen in our patient. Turner syndrome is classically
associated with elevated FSH and LH, a result of hypergonadotophic
hypogonadism secondary to primary ovarian failure. A child, whose
clinical phenotype and karyotype suggestive of Turner syndrome with
normal/low FSH should be evaluated for an associated primary defect in
the pituitary/hypothalamus. Elevated anti-thyroid peroxidase antibody
titer in our patient is reflective of underlying thyroid autoimmunity.
Turner syndrome is well known to be associated with increased occurrence
of various autoimmune disorders, which include autoimmune thyroid
disorders and inflammatory bowel disease [1].
Effect of GH in Turner syndrome is inferior to that
in cases of isolated GH deficiency [8]. Factors associated with improved
final height in Turner syndrome include higher baseline height, higher
mid parental height, higher GH dose used, younger age at GH initiation
and longer therapy [1]. Levothyroxine and sex steroids may have
contributed to the better height outcome seen in our patient.
Indications for termination of GH in Turner syndrome include bone age 14
years or growth velocity <2 cm/year [9,10]. Recently it has been
suggested that early initiation of GH in Turner syndrome before onset of
growth failure (before 4 years age), results in 80% children achieving
normal stature [3]. Also it has been suggested that early initiation of
very low dose estradiol (at 5 years of age instead of at 12 years),
along with GH results in additional increase in adult stature by 2.1 cm
[10].
Limitations of this report include lack of genetic
testing for transcription factor defects causing MPHD. However, most
transcription factor defects present in early childhood, except for
PROP1 mutation [2]. Clinical profile of our patient did not match with
that of any known transcription factor defect. We conclude that any
Turner syndrome patient, with height <3 rd
percentile on TSGC, should be further evaluated for additional
conditions.
Contributors: DD, CS: clinical and
investigational work-up of the patient; SM, DD: patient management. All
authors contributed to the preparation of the manuscript.
Funding: None; Competing interests:
None stated.
References
1. Pinsker JE. Clinical review: Turner syndrome:
Updating the paradigm of clinical care. J Clin Endocrinol Metab.
2012;97:E994-1003.
2. Dutta D, Roy A, Ghosh S, Mukhopadhyay P, Dasgupta
R, Mukhopadhyay S, et al. Rathke’s cyst with ectopic
neurohypophysis presenting as severe short stature with delayed puberty.
Indian J Endocrinol Metab. 2012;16:S288-90.
3. Linglart A, Cabrol S, Berlier P, Stuckens C,
Wagner K, de Kerdanet M, et al. Growth hormone treatment before
the age of 4 years prevents short stature in young girls with Turner
syndrome. Eur J Endocrinol. 2011;164:891-7.
4. Valenta LJ, Elias AN, Bocian M. Atypical
biochemical findings in Turner’s syndrome: Identification of a possible
subset. Fertil Steril. 1984;42:798-802.
5. Afonso-Lopes L, Benador D, Wacker P, Wyss M, Sizonenko
PC. Turner’s syndrome and hypogonado-trophic hypogonadism: thalassemia
major and hemo-chromatosis. J Pediatr Endocrinol Metab. 1995;8:73-7.
6. Donti E, Nicoletti I, Venti G, Filipponi P, Gerli
R, Spinozzi F, et al. X-ring Turner’s syndrome with combined
immunodeficiency and selective gonadotropin defect. J Endocrinol Invest.
1989;12:257-63.
7. Efstathiadou Z, Tsatsoulis A. Turner’s syndrome
with concomitant hypopituitarism: Case report. Hum Reprod.
2000;15:2388-9.
8. Stephure DK. Impact of growth hormone
supplementation on adult height in Turner syndrome: Results of the
Canadian randomized controlled trial. J Clin Endocrinol Metab.
2005;90:3360-6.
9. Van-Pareren YK, de Muinck-Keizer-Schrama SM,
Stijnen T, Sas TC, Jansen M, Otten BJ, et al. Final height in
girls with Turner syndrome after long-term growth hormone treatment in
three dosages and low dose estrogens. J Clin Endocrinol Metab.
2003;88:1119-25.
10. Cuttler L, Rosenfield RL. Assessing the value of treatments to
increase height. N Engl J Med. 2011; 364:1274-6.
|
|
|
 |
|
