|
|
|
Indian Pediatr 2011;48: 731-732 |
 |
Wiedemann-Rautenstauch Syndrome |
|
JP Narayan, P Garg, G Pareek and *S Narayan
From the Departments of Pediatrics and *Obstetrics, JLN
Medical College, Ajmer, Rajasthan, India.
Correspondence to: Dr Jaiprakash Narayan, C/o Shri
Rajendra Prasad, 123/12 Agrawal Farm Thadi Market Mansarowar, Jaipur,
Rajasthan 302 020.
Email:
[email protected]
Received: January 22, 2010;
Initial review: March 25, 2010;
Accepted: June 28, 2010.
|
Wiedemann-Rautenstauch (WR) syndrome is a rare autosomal recessive
neonatal progeroid syndrome with only few published case reports. We
describe a neonate showing clinical features of WR syndrome with
peeling of skin, and presented with weak cry and breathing difficulty
since birth.
Key words: Neonate, Progeria, Wiedemann-Rantenstrauch
syndrome.
|
|
Wiedemann-Rautenstauch (WR) syndrome is a known neonatal progeroid
syndrome comprising of generalized lipoatrophy except for fat pads
in the suprabuttock areas, hypotrichosis of the scalp hair, eyebrows
and eyelashes, relative macrocephaly and macroglosia [1]. Till date,
total 34 cases have been reported and none from India. [2-9].
Case Report
This newborn infant, delivered in a district
hospital, was admitted with complaints of weak cry and breathing
difficulty since birth. She was the first daughter of healthy
non-consanguineous 23-year-old mother and 27-year-old father.
Delivery was normal at 36 weeks of gestation and birthweight was 1.5
kg, length 43 cm and occipito-frontal head circumference was 34 cm.
There was no history of birth of similar children in family and in
close relatives. No significant antenatal history was present and
baby died on third day of an undetermined cause. Physical
examination at the time of admission showed apparent growth retarded
baby with macrocephaly with frontal and biparietal bossing,
craniofacial disproportion, almost total alopecia, large fontanels
and wide sutures, prominent scalp veins, hypoplasia of facial bones,
small nose, upward slanting palpebral fissures, hyperteleorism,
ocular proptosis, sparse eye-brows and eyelashes, low set and small
ears with normal configuration, down turned angle of mouth, long
filtrum, high arched palate, and a sharp and pointed chin (Fig.1).
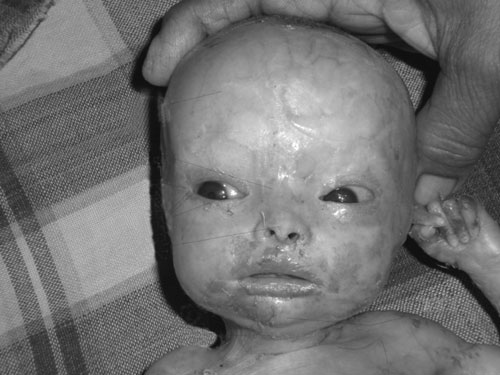 |
|
Fig. 1 Macrocephaly with
craniofacial disproportion, total alopecia, hypoplasia of
facial bones, upward slanting palpebral fissures,
hypertelorism, sparse eyebrows and eyelashes, and downturned
angle of mouth. |
The neck was short with redundant skinfolds,
nipples wide spaced and there was no cardiac murmur and air entry
was bilaterally equal and normal. Abdomen was slightly distended.
Liver and spleen were palpable. The external genitalia were of a
normal female. There was generalized deficient subcutaneous fat,
with the exception of excessive fat on the buttocks. The skin was
thin, shiny, erythematous and there was peeling of skin. Fingers and
toenails were normal. She was hypertonic. A complete blood count
showed Hb 15.6 g/dL, TLC 9800, DLC N-65%, L-28%, blood sugar,
calcium were normal, X–ray showed bilateral infiltration, and USG
cranium was normal. Karyotyping was not sent because parents refused
for it. Above clinical findings confirmed a clinical diagnosis of
typical WR progeroid syndrome.
Discussion
The clinical features of our case are similar to
those described earlier [2-9]. Patients with this syndrome can be
recognized at birth because of distinct clinical features that
include short stature, failure to thrive, progeroid appearance,
apparent macrocephaly with frontal and parietal bossing, wide
fontanels and sutures, prominent scalp veins, hypoplasia of facial
bones, sparse scalp hair, eyebrows and eyelashes, and generalized
lipoatrophy. Most patients showed neonatal teeth, which were lost
early [6]. These patients also have endocrine abnormalities such as
hypertriglyceridemia, hyper-cholesterolemia and hyperinsulinemia but
not required for diagnostic purpose [3]. Majority of these patients
die during the first few days or months after birth [6]. At present,
there is no treatment.
Contributors: JP and GP: patient history
taking and management, and review of literature. SN: review of
literature and manuscript writing. PG supervised the management and
drafted the manuscript. JP shall act as a guarantor.
Funding: None.
Competing interests: None stated.
References
1. Pivnick EK, Angle B, Kaufman RA, Hall BD,
Pitukcheewanout P, Hersh JH, et al. Neonatal progeroid (Wiedemann-
Rautenstrauch) syndrome: report of five new cases and review. Am J
Med Genet. 2000;90:131-40.
2. Arboleda H, Arboleda G. Follow-up study of
Wiedemann-Rautenstrauch syndrome: Long-term survival and comparison
with Rautenstrauch’s patient "G". Birth Defects Res. 2005;73:562-8.
3. Morales LC, Arboleda G, Rodriguez Y, Forero
DA, Ramirez N,Yunis JJ, et al. Absense of lamin A/C gene
mutations in four Wiedemann-Rautenstrauch syndrome patients. Am J
Med Genet. 2009;149A:2695-9.
4. Wiedemann HR. An unidentified neonatal
progeroid syndrome: follow-up report. Eur J Pediatr. 1979;130:65-70.
5. Rautenstrauch T, Snigula F. Progeria: a cell
culture study and clinical report of familial incidence. Eur J
Pediatr. 1977;124:101-11.
6. Arboleda H, Quintero L, Yunis E.
Wiedemann-Rautenstrauch neonatal progeroid syndrome: report of three
new patients. J Med Genet. 1997;34:433-37.
7. Singer A, Devriendt K, Dev D, Vinkler C. A
patient with neonatal progeroid (Wiedemann-Rautenstrauch) syndrome
and complex chrosomal rearrangement (CCR). Available from:
www.ashg.org/2009meeting/abstracts/fulltext/f20712.htm /. Accessed
May 1, 2010.
8. Dinleysi EC, Tekin N, Dinleyici M, Aksit MA.
Clinical and laboratory findings of two newborns with
Wiedemann-Rautenstrauch syndrome: additional features, evaluation of
bone turnover and review of literature. J Pediatr Endocrinol Metab.
2008;21:591-6.
9. Tunc T, Bulbul A, Erdinc K, Sarici SU, Gul D,
Ozcan O. The Wiedemann-Rautenstrauch or neonatal progeroid syndrome:
report of a patient with hypospadias. Genet Couns. 2009;20:367-71.
|
|
|
 |
|
