Griscelli syndrome is a rare autosomal recessive
disease characterised by pigmentary dilution of skin and hair, variable
cellular immunodeficiency and an acute phase of uncontrolled T
lymphocyte and macrophage activation leading to fatal hemophagocytic
syndrome. It was first described by Griscelli in 1978, and since then
only around 60 cases have been reported, mostly from the Turkish and
Mediterranean population. To the best of our knowledge this is the first
case reported from India, which has been proven by mutation syudy.
Case Report
A 13-day-old male baby born of second degree
consanguineous parentage was brought for evaluation of excessively fair
skin and silvery gray hair. His birth weight was 3.25 kg. The mother had
conceived five times, resulting in one abortion, a normal female child
who is now 4 years old, and two elder male siblings who had the same
clinical features as the proband and in addition had photophobia and
delay in motor and mental milestones. They both expired at around 15
months of age after developing hepatosplenomegaly, neutropenia, seizures
and developmental regression. One child was investigated in detail for
metabolic problems and his peripheral smear failed to show any abnormal
granules in the granulo-cytes. There was no history of any relatives
affected by similar clinical presentation.
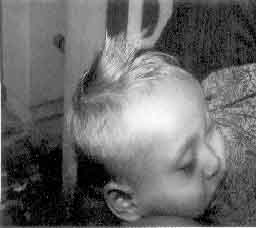
On examination the child had generalized excessively
fair skin when compared with his parents and living sibling. He had
silvery gray scalp hair, white eyelashes, eyebrows and body hair (
Fig. 1). All other systems were within normal limits.
 |
|
Fig. 1. Typical silvery gray hair and
hypopigmented skin of the proband at 9 months |
The possibility of Chediak-Higashi Syndrome (CHS) was
considered, with the two siblings dying due to the "accelerated phase".
Peripheral smear of the proband failed to show any abnormal granules
even after repeated examination. Intracytoplasmic granules were absent
on the peripheral smear of one of the elder siblings which was examined
while he was terminally ill from old records. Serum copper and
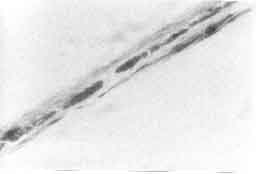
ceruloplasmin levels were normal. Microscopic examination of the hair
shaft of the proband showed uneven aggregations of large pigment
granules, and hypopigmentation in the rest of the hair which is the
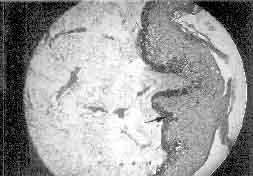
hallmark of Griscelli syndrome (Fig. 2). Light microscopy of skin
showed large clumps of melanin granules in melanocytes in the basal cell
layer in Masson-Fontana stained sections, which is consistent with the
skin changes of Griscelli Syndrome (Fig. 3).
 |
 |
|
Fig. 2. Microscopic examination of hair showing uneven
aggregation of large pigment granules (× 400). |
Fig. 3. Skin biopsy showing large clumps of melanin granules
in the basal cell layer (Masson-Fontana × 400) |
Mutation study of the proband and the parents were
done at Hospital Necker-Enfants Malades, Paris. Rab27a gene was
sequenced from the proband and a homozygous C550T leading to R184X
mutation (X is a stop mutation) in Rab27a gene was
identified.This nonsense mutation is the cause for Griscelli syndrome.
Both parents were found to be heterozygous carriers of the same
mutation.
The child is on regular follow up. He is 1-year-old
now and weighs 9 kg and has normal motor and mental development for his
age. He has not yet developed any features of the "accelerated phase".
Bone marrow transplantation is being considered.
Discussion
Griscelli Syndrome (GS) is an extremely rare
autosomal recessive disorder characterized by partial albinism with
silvery gray hair, eyebrows and eyelashes, in association with cellular
immunodeficiency, frequent infections, and neurological abnormalities. A
fatal outcome occurs in the so-called "accelerated phase" of the
disorder, which is caused by uncontrolled T lymphocyte and macrophage
activation(1). Thus it is clinically similar to Chediak Higashi
Syndrome.
The single most consistent cutaneous expression of
albinism in these patients is silvery gray sheen to their hair(2).
Microscopic examination of the hair shaft provides strong support for
the diagnosis of these syndromes, and allows one to distinguish between
them. In both syndromes the hair shaft contains a typical pattern of
uneven accumulation of large pigment granules, instead of the
homogeneous distribution of small pigment granules seen in normal hair.
In GS the clusters of melanin pigment on the hair shaft are six times
larger than in CHS.
The hallmark of CHS is the presence of giant
intracytoplasmic granules in virtually all granulated cells, which is
never observed in GS. Light microscopy of the skin in GS shows
hyperpigmented melanocytes with poorly pigmented adjacent keratinocytes,
instead of the homogeneous distribution of melanin granules in
melanocytes and keratinocytes as seen in normal epidermis(3).
The disease is mapped on chromosome 15q21 locus and
mutations of MyoVa or Rab27a gene can lead to GS. Although pigmentary
dilution is identical in both groups, only patients with Rab27a
mutations have abnormal lymphocyte cytotoxic activity which result in
haemophagocytic syndrome. Severe neurological impairment with no immune
deficiency is the characteristic feature with MyoVa mutation as this
gene regulates organelle transport in melanocytes and in neuronal cells.
Impairment of intracellular trafficking and secretion of several
lysosomal proteins including melanin from melanocytes and the lytic
enzymes from cytotoxic cells occur in GS and CHS. The secretory defect
accounts for the hypopigmentation and the cellular immunodeficiency(4).
The immuno-logic abnormalities are restricted to the patients with
Rab27a mutation as the capacity of the lymphocytes and NK cells of these
patients to lyse target cells is impaired or absent, due to a consistent
inability to secrete cytotoxic granules. MyoVa defect does not affect
cytotoxic granule secretion and hence they never develop accelerated
phase.
Elejalde syndrome or Melanolysosomal neuroectodermal
syndrome is another rare autosomal recessive disorder with striking
resemblance to GS and they manifest with hypopigmentation, silvery hair
and early onset severe psychomotor retardation without immne deficiency
strongly suggesting that Elejalde syndrome and GS with MyoVa mutation
are allelic(5). Griscelli, Chediak Higashi and Elejalde syndromes are
compared in Table I.
TABLE I
Comparison of Griscelli, Chediak-Higashi and Elejalde Syndromes.
| |
Griscelli
syndrome |
Chediak-Higashi
syndrome |
Elejalde
syndrome |
Hypopigmentation
|
+
|
+
|
+
|
Silvery hair
|
+
|
+
|
+
|
Immune deficiency
|
+
|
+
|
–
|
Neurological impairment
|
+ MyoVa
– Rab27a
|
–
|
++
|
Intracytoplasmic granules
|
–
|
+
|
–
|
Clusters of melanin pigments in hair microscopy
|
++
|
+
|
++
|
Impaired transfer of melanin to keratinocytes in skin biopsy
|
+
|
+
|
+
|
Mode of inheritance
|
AR
|
AR
|
AR
|
Same mutation in Rab27a as the one observed in the
proband reported here was previously identified in Turkish, Italian,
Danish, English and Mauritius patients but ours is the first patient
analysed from India (6).
The prognosis for long term survival in GS due to
Rab27a defect is relatively poor. This disorder is rapidly fatal during
the accelerated phase of the disease. Etoposide was found to be
effective in some cases during the accelerated phase(7). Antithymocyte
globulin and cyclosporin A have also achieved remission in a few
cases(8). Allogenic bone marrow transplantation is the only curative
treat-ment(9). In MyoVa defect the neurological impairment and
psychomotor delay do not improve and hence there is no role for bone
marrow transplantation.
Acknowledgements
The authors would like to thank Dr. Genevieve de
Saint Basile of Hospital Necker-Enfants Malades, Paris who provided
information and did the mutation study for confirming the diagnosis.
Contributors: SSR diagnosed and manages this
case; she will act as guarantor of the paper. LM and SJI were jointly
involved in the interpretation of histopathology.
Funding: None.
Competing interests: None.
