Sriparna Basu
Dilip Kumar Paul
Somprakas Basu
Bengal
Medical College and Hospitals, Sushrutnagar, Darjeeling, West Bengal,
India.
Correspondence to: Dr.
Sriparna Basu, 113, Ultadanga Main Road, Kolkata-700 067, West Bengal,
India.
E-mail: [email protected]
Manuscript received: June
20, 2001;
Initial review completed:
August 3, 2001;
Revision accepted: April 22, 2002.
expression in the
parents. The rarity of the disease is highlighted and the intragroup
variations are discussed.
The Hereditary Sensory and Autonomic
Neuropathies (HSAN) are a group of rare disorders characterized by
prominent sensory and autonomic neuropathy without motor involvement(1).
They reflect failure of development or degeneration of sub-populations
of peripheral sensory and autonomic neurons. Classification is done into
five main groups based on inheritance, clinical features and the
population of sensory neurons affected. Impaired pain appreciation
results in mutilating acropathy with skin ulceration and fissuring, long
bone fractures, Charcot’s joints and digit amputation. The precise
symptoms and signs and the nerve conduction abnormalities of each type
are determined by the subpopulation of sensory neurons predominantly
affected(2). We report a family in which all four siblings were affected
with HSAN Type II without any
Case Report
Four children (siblings),
two males and two females presented with history of insensitivity to
pain and temperature and multiple painless ulcerations over various
parts of the body since infancy. They were born of consanguineous
marriage in a Muslim family. All were of full term normal vaginal
delivery at home with uneventful antenatal, intranatal and postnatal
period. There was no history of recurrent episodes of fever, bladder and
bowel habits were normal and the immunization history was complete. The
course of the disease was slowly progressive, the eldest siblings being
most affected. Both parents were phenotypically normal. There was no
history of abortion or stillbirth and no family history of such illness
in either of the parents’ side.
Examination revealed generalized loss of
all modalities of sensation, more marked on the distal parts of the
limbs and cornea. Multiple ulcerations were present on the hands, feet,
lips and alae of the nose. All showed mutilating acropathy in the form
of complete and/or partial loss of digits and foot deformities (Fig. 1).
Other features included anhidrosis over distal parts of limbs, palmer
and planter callosities, corneal opacities (cases 1 and 2), bowing of
legs with radiological evidence of rickets (cases 2 and 3) (Table I).
There was no sign of peripheral nerve thickening. Except total areflexia
(superficial and deep), motor functions were well preserved. Fundus
examination, rest of the central nervous system and other systemic
examinations including blood pressure were normal. Anthropometric
measurements as per Indian Council of Medical Research standard(3) and developmental milestones were within normal limits.
Besides high serum alkaline phosphatase (> 1000 IU/L), routine
hematological investigations were normal. Detailed biochemical
assessment was done in all. Serum calcium, blood urea and creatinine
were within normal limits while serum phosphorus was slightly below
normal. Based on the above features a provisional clinical diagnosis of
HSAN was made and electrophysiological studies and nerve biopsies were
performed in all the siblings to confirm the diagnosis. Motor nerve
conduction veloctiy (MNVC) studies were done on the median and ulnar
nerves on both sides, with a stimulus of 3mV, 63mA, sweep of 2msec,
duration of 0.1 sec and recording distance of 140 mm. Result was
interpreted by computerized graphical representation. The mean latency
difference was 2.84 milliseconds (range 2.40 - 3.68) and mean conduction
velocity (CV) was 57.69 meters/sec (range 51.47 - 62.50). The sensory
nerve conduction velocity (SNVC) studies were done by ring electrodes on
the digital nerves of the upper extremity by the principle of
orthodromic conduction. All showed non-recordable sensory conduction.
NCV studies were not done on the parents as they were phenotypically
normal and showed no evidence to indicate sensory neuropathy. Sural
nerve biopsies showed complete absence of myelination and no evidence of
Schwann cell proliferation.
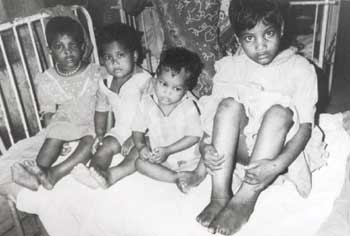
Fig. 1. Siblings
showing mutilating acropathy.
The cases were managed conservatively.
Infected ulcers were treated with antibiotics and antiseptic dressings.
Proper foot and skin care was ensured and adequate counselling was done
to prevent trauma and improve personal hygiene to prevent infections.
Rickets (overt and subclinical) was treated with a single intramuscular
injection of 6,00,000 IU of vitamin D and oral calcium supplementation.
After 3 weeks, biochemical parameters were reassessed. A definite
decrease was seen in serum alkaline phosphatase levels indicating
response to megadose vitamin D therapy. Serum phosphorus levels showed
improvement and radiological
signs of healing rickets were seen in all.
Discussion
According to the
descriptive classification of Donaghy et al(4) HSAN type-I or hereditary
sensory radicular neuropathy is dominantly inherited. Symptoms begin in
second or later decades with sensory loss and subsequent tissue injury.
Pain is more affected than touch. Sural nerve biopsy shows loss of
unmyelinated fibres more than the myelinated ones. Type-II HSAN is
recessively inherited and begins in infancy. There is generalized
pansensory loss. Autonomic disturbances include bladder dysfunction,
impotence and anhidrosis of hands and feet. Motor function is preserved
but tendon reflexes are usually lost. Association of retinitis
pigmentosa, motor weakness and neurotrophic keratitis with sensory
neuropathy has been described(1,2). Sensory nerve action potentials are
absent and there is loss of myelinated fibres in sural nerve biopsy(1).
Types-III, IV and V are also autosomal recessive and type-III is
associated with prominent autonomic involvement(1,5). Nerve biopsy shows
reduced number of unmyelinated fibres. Type-IV is associated with bouts
of pyrexia, failure to thrive, anhidrosis and mental retardation.
Clinical manifestations of Type-V include mutilating acropathy and
bilateral neurotrophic keratitis. Motor functions and tendon reflexes
are normal. Sural nerve biopsy shows selective reduction of smaller
myelinated nerve fibre population(2).
Table - Comparison of Characteristics of the Patients
|
Case 1
|
Case 2
|
Case 3
|
Case 4
|
Age (years)
|
9
|
7
|
6
|
4
|
Sex
|
F
|
F
|
M
|
M
|
Age of onset
|
Infancy
|
Infancy
|
Infancy
|
Infancy
|
Skin ulceration
|
+
|
+
|
+
|
+
|
Mutilating acropathy
|
+
|
+
|
+
|
+
|
Motor function
|
Preserved
|
Preserved
|
Preserved
|
Preserved
|
Tendon reflexes
|
–
|
–
|
–
|
–
|
Distal anhidrosis
|
+
|
+
|
+
|
+
|
Bladder & bowel habits
|
N
|
N
|
N
|
N
|
Corneal opacity
|
+
|
+
|
–
|
–
|
Fundus examination
|
N
|
N
|
N
|
N
|
Clinical and radiological rickets
|
–
|
+
|
+
|
–
|
Serum alkaline phosphatase
|
|
|
|
|
Sensory nerve conduction velocity
|
No AP
|
No AP
|
No AP
|
No AP
|
Motor nerve conduction velocity
|
N
|
N
|
N
|
N
|
Sural nerve biopsy
|
Complete absence of myelinated nerve fibres and
no evidence of schwann cell proliferation
|
AP = Action Potential
In view of the history of
consanguinity, autosomal recessive transmission was likely in our
patients. Clinical features, nerve conduction velocity studies and sural
nerve biopsies of all the siblings correlate well with those of type-II
variety of HSAN(1,2,6). A similar form of HSAN may be inherited as
x-linked recessive trait(7). The usual autonomic disturbances in the
form of bladder and bowel involvement and hypertension were absent in
our cases, though distal anhidrosis was present uniformly in all. In
this regard it is highlighted that anhidrosis is a prominent
manifestation of type-IV variety. Motor functions were well preserved.
Association of spastic paraplegia with HSAN type-II has been reported by
Cavanagh et al(8) though such feature was absent in our cases.
Uniformity in the
presence or absence of clinical features was seen in all except for
corneal opacities and clinical and radiological evidence of rickets.
Only 2 cases showed corneal opacities. Though serum alkaline phosphatase
was raised in all of them, two had bowing of legs and classical
radiological features of rickets (Table I). Since the biochemical
profile indicated no abnormality of renal function the cause of rickets
was most likely nutritional and probably coincidental as also supported
by the response to megadose vitamin D therapy. However, in light of
anthropometric measurements the nutrition of the children was good and
no evidence of clinical nutritional deficiency was seen except overt
rickets in only two of them. HSAN is a rare disorder. In India,
Balachandran et al(6) has reported type-II variety in siblings. Initial
delay in diagnosis can occur due to similarity of the mutilating
acropathy with that of leprosy, a more common disease in India. Hence
the latter should always be excluded prior to diagnosis of this rare
disorder.
All the described
features may not be found in a single case of HSAN. Intragroup
variations are present and are still being reported(9,10). The spectrum
of clinical features, nerve conduction studies, inheritance patterns and
nerve biopsies form the mainstay of diagnosis and subtyping. Treatment
is usually symptomatic. Prevention of trauma to the anesthetized parts
and appropriate management of the sequele of autonomic neuropathy are
the aims. Prognosis is guarded as adequate long term follow up series
are lacking(1,4).
Contributors: SB and SPB
did the clinical work up and drafted the paper. DKP coordinated the case
management and supervised the drafting. SB will act as the guarantor for
the paper.
Funding: None.
Competing interests: None stated.
|
Key
Messages |
| •
Diagnosis of HSAN type-II was based on pansensory loss, mutilating acropathy,
corneal anesthesia and total areflexia with normal motor function. Though
bladder and bowel functions and blood pressure were normal, distal
anhidrosis was seen. Sensory nerve conduction was absent and sural nerve
biopsy showed loss of myelination. |
