dentification of a carrier of balanced
translocation is important as the risk of imbalanced gametes in the
translocation carrier is significant and can lead to recurrent abortions
or malformed babies [1]. Similarly, there may be some more balanced
carriers in the family, who need to be detected to provide proper
genetic counseling.
Traditional karyotype is the gold standard for
detection of balanced chromosomal translocations [2]. In many cases,
karyotype fails to detect chromosomal translocations, especially when
the translocation does not change the band pattern or the length of the
chromosomes. We report a unique cryptic balanced translocation in a
family segregating for at least three generations leading to unbalanced
offspring that could not be identified by traditional karyotype and was
revealed after cytogenetic microarray (CMA) in two offspring.
Case Report
Case 1
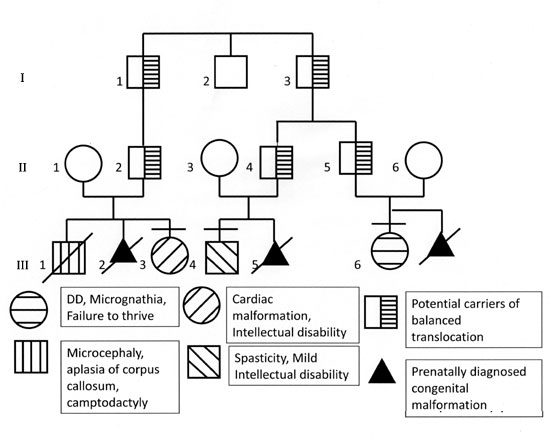
Proband (III-3 in the pedigree, Fig. 1)
was referred at 5 months of age because of congenital heart disease and
facial dysmorphism. On examination, weight was 3.6 kg (-4 SD for 5
months), length was 55.5 cm (-3 SD for 5 months) and head circumference
was 41 cm (50th centile for 5 months). She had bi-temporal narrowing,
eversion of the right lower eye lid and thin lower lip. Both thumbs and
great toes were broad. She had bilateral single palmar crease and
camptodactyly in both the hands. The proband was lost to follow up and
was re-evaluated at 5 years. She achieved neck holding at 6 months,
sitting without support at 2 years and standing without support at 4½
years. She responded to her name and stranger anxiety was present since
3 years of age. She started saying bisyllables at 2 years. Conventional
cytogenetic analysis with the G banded karyotype (at 550 band level) and
magnetic resonance imaging (MRI) brain were normal. Echocardiography
demonstrated atrial septal defect with patent ductus arteriosus.
Cytogenetic microarray (CMA) result using Affymetrix 2.7 M array (Santa
Clara, CA, USA) showed 9.4 Mb gain of 7q36.1-36.3 and 13.16 Mb loss of
chromosome 11q24.1-25 [arr7q36.1q36.3(149698257-159118443) ×3,
11q24.1-25(121769912-134926021] X1.
 |
|
Fig. 1 Pedigree of the family.
|
Mother of the proband (II-1) had the history of
prenatally detected congenital malformations in the previous two
pregnancies. First baby (III-1 in Fig.1) was a male who
had microcephaly, corpus callosal defect and some congenital anomalies
of the lung. The baby expired 3 hours after birth. Second pregnancy was
terminated at 18 weeks as the fetus was diagnosed to have alobar
holoprosencephaly with microcephaly.
Case 2
This child (III-6) was a 4 month old female, first
born child of a non consanguineous couple. Her father was the cousin of
the father of case I. The child had feeding problems and was not growing
well since birth. She was delivered post term with a birth weight of 2.1
kg. During the antenatal period maternal serum screening test was
positive for trisomy 21. Amniocentesis was performed and karyotype at
550 band level was normal. There was a history of delayed cry and
respiratory distress at birth.
On examination, the child’s weight was 2.7 kg (–7 to
–6 SD for 4 months), length was 50 cm (–5 SD for 4 months) and head
circumference was 32 cm (–6 SD for 4 months). Facial dysmorphism
included low set ears, broad nose, left eye ptosis, high arched palate
and microretrognathia (Fig. 2B). The nails in lower limbs
were hypoplastic. There was no camptodactyly. MRI brain revealed corpus
callosum agenesis and hypoplastic inferior vermis. The baby expired at 8
months. CMA analysis showed 13 Mb gain of genomic material on 11q 24.1
-25 along with 9.2 Mb loss on 7q 36.1 – 36.3 region. (arr7q36.1q36.3
(149770238-159118443) X 1, 11q24.1-25 (121769912-134926021) X 3.
Another child in the family (III-4 in Fig.
1) was also found to have some developmental disability and he was
related through his father. There was a history of intellectual
disability with spasticity in him. He had porencephaly on MRI brain and
the cytogenetic microarray revealed normal results. In the wife of the
same paternal uncle, one pregnancy was terminated after prenatal
diagnosis of holoprosencephaly (III-5).
The mother of III-6 (II-6 in Fig. 1)
returned in her second pregnancy for prenatal diagnosis. Chorionic
villus sampling was done at 11 weeks and Multiplex ligation probe
amplification revealed same genomic imbalance involving the terminal
regions of chromosomes 7 and 11 (loss at 7qter and gain at 11qter) as in
the proband III-6. They decided to terminate the pregnancy and the fetal
autopsy did not reveal any major or minor malformation.
The unbalanced genomic rearrangements in the two
cousins involving the same breakpoints suggest that the father of the
proband and also of her cousin may be carriers of balanced
translocation. To confirm this, we performed Fluorescent in Situ
Hybridisation (FISH) analysis using centromeric probes for chromosome 7
and 11 and probes for subtelomeric region chromosome 7q36 and 11q24.1.25
in the father [II-2] of III-3. FISH confirmed the translocation between
chromosomes 7 and 11.
Discussion
In this family the possibility of balanced
translocation in the fathers was confirmed by FISH in one of the
possible carriers (II-2). The other possible carrier II- 5 though not
confirmed by FISH, was an obligate carrier as his daughter had
imbalances of the same chromosomes with the same breakpoints. II-4 is
also likely to be the carrier of same translocation, as his wife’s one
pregnancy showed a fetus with holoprosencephaly, but CMA of the fetal
sample and FISH for II 4 was not done and his son with a different
phenotype did not show any genomic imbalance.
This case highlights the utility of cytogenetic
microarray in cases with normal karyotype and most importantly the
possibility of familial balanced translocation in cases of double
segment imbalances. It is important to identify translocation carriers
as they have a high risk of conceptions with genomic imbalances and can
be helped by prenatal diagnosis.
There are a number of genes present in the deleted
and duplicated region involving chromosome 7q36.1 and 11q24.1. The
relevant genes on chromosomes 7(q36.1-36.3) and 11(q24.1-25) include two
genes for holoprosencephaly (HPE), namely SHH (Sonic Hedge hog)
at 7q36 and CDON (cell adhesion molecule-related downregulated by
oncogenes) at 11q24 which explains the malformation in the fetuses. The
penetrance of holoprosencephaly genes is 70% [3] which explains the
absence of holoprosencephaly in our probands. KIRREL 3 gene
responsible for mental retardation is present in the region ch
11q24.1-25 which explains the mental retardation in the 2 cousins.
A large study involving 5380 patients identified the
chromosomal abnormalities in subtelomeric region detected by microarray
[4]. There are a number of case reports related to the double segment
imbalances detected by cytogenetic microarray [5,6]. This case stresses
the need to suspect chromosomal abnormality in families with multiple
members affected with different phenotypes of intellectual disability
and dysmorphism.
1. Tilak P. Effect of reciprocal translocations on
phenotypic Abnormalities. Int J Hum Genet. 2010;10:113-9.
2. Nicolini U, Lalatta F, Natacci F, Curcio C, Bui
TH. The introduction of QF-PCR in prenatal diagnosis of fetal
aneuploidies: time for reconsideration. Hum Reprod Update.
2004;10:541-8.
3. Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi
DW, Die-Smulders C, et al. The mutational spectrum of the sonic
hedgehog gene in holoprosencephaly: SHH mutations cause a significant
proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;
8:2479-88.
4. Shao L, Shaw AC, Lu XY, Sahoo T, Carlos AB, Lalani
SR, et al. Identification of chromosome abnormalities in
subtelomeric regions by microarray analysis: a study of 5,380 cases. Am
J Med Genet A. 2008;146:2242-51
5. Petriczko E, Biczysko-Mokosa A, Bogdanowicz J,
Constantinou M, Zdziennicka E, Horodnicka-Jozwa A, et al.
Familial distal monosomy 3p26.3-pter with trisomy 4q32.2-qter,
presenting with progressive ataxia, intellectual disability, and
dysmorphic features. Am J Med Genet A. 2012;158:1442-6.
6. Shojaei A, Behjati F, Derakhshandeh-Peykar P,
Razzaghy-Azar M, Otukesh H, Kariminejad R, et al. Partial trisomy
7q and monosomy 13q in a child with disorder of sex development:
phenotypic and genotypic findings. Gene. 2013;517:137-45.


