1. Nio M, Ohi R, Miyano T, Saeki M, Shiraki K,
Tanaka K, Japanese Biliary Atresia Registry. Five- and 10-year
survival rates after surgery for biliary atresia: A report from the
Japanese Biliary Atresia Registry. J Pediatr Surg. 2003;38:997-1000.
2. McKiernan PJ, Baker AJ, Kelly DA. The
frequency and outcome of biliary atresia in the UK and Ireland.
Lancet. 2000;355:25-9.
3. Davenport M, Tizzard SA, Underhill J,
Mieli-Vergani G, Portmann B, Hadzic N. The biliary atresia splenic
malformation syndrome: A 28-year single-center retrospective study.
J Pediatr. 2006;149:393-400.
4. Narasimhan KL, Chowdhry SK, Vaiphei K, Samujh
R, Mahajan JK, Thapa BR, et al. Outcome of biliary atresia
from Chandigarh: Results of a prospective analysis. Indian Pediatr.
2001;38:1144-8.
5. Yachha SK. Cholestatic jaundice during
infancy. Indian J Gastroenterol. 2005;24:47-8.
6. Hsiao CH, Chang MH, Chen HL, Lee HC, Wu TC,
Lin CC, et al. Universal screening for biliary atresia using
an infant stool color card in Taiwan. Hepatology. 2008;47:1233-40.
7. Gu YH, Yokoyama K, Mizuta K, Tsuchioka T, Kudo
T, Sasaki H, et al. Stool color card screening for early
detection of biliary atresia and long-term native liver survival: A
19-year cohort study in Japan. J Pediatr. 2015;166:897-902.
8. Tang KS, Huang LT, Huang YH, Lai CY, Wu CH,
Wang SM, et al. Gamma-glutamyl transferase in the diagnosis
of biliary atresia. Acta Paediatr Taiwan. 2007;48:196-200.
9. Humphrey TM, Stringer MD. Biliary atresia: US
diagnosis. Radiology. 2007;244:845-51.
10. Kianifar HR, Tehranian S, Shojaei P,
Adinehpoor Z, Sadeghi R, Kakhki VR, et al. Accuracy of
hepatobiliary scintigraphy for differentiation of neonatal hepatitis
from biliary atresia: Systematic review and meta-analysis of the
literature. Pediatr Radiol. 2013;43:905-19.
11. Yachha SK, Khanduri A, Kumar M, Sikora SS,
Saxena R, Gupta RK, et al. Neonatal cholestasis syndrome: An
appraisal at a tertiary center. Indian Pediatr. 1996;33:729-34.
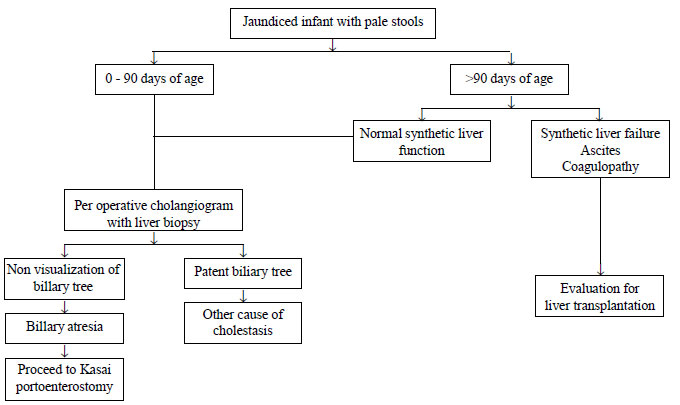
12. Poddar U, Thapa BR, Das A, Bhattacharya A,
Rao KL, Singh K. Neonatal cholestasis: Differentiation of biliary
atresia from neonatal hepatitis in a developing country. Acta
Paediatr. 2009;98:1260-4.
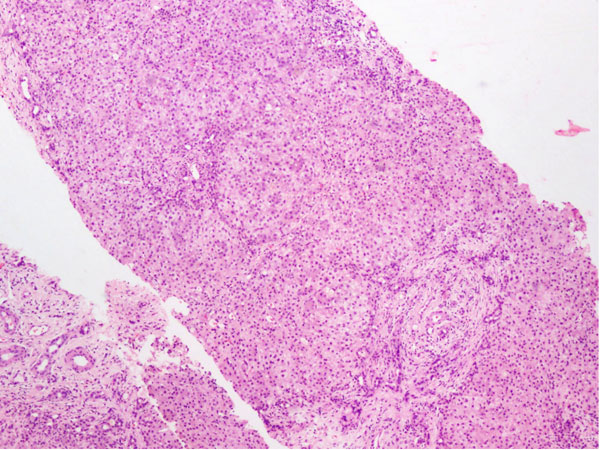
13. Rastogi A, Krishnani N, Yachha SK, Khanna V,
Poddar U, Lal R. Histopathological features and accuracy for
diagnosing biliary atresia by prelaparotomy liver biopsy in
developing countries. J Gastroenterol Hepatol. 2009;24:97-102.
14. Indian Academy of Pediatrics. Pediatric
Gastroenterology Subspecialty Chapter. Consensus report on neonatal
cholestasis syndrome. Indian Pediatr. 2000;37:845-51.
15. Davenport M, Betalli P, D’Antiga L, Cheeseman
P, Mieli-Vergani G, Howard ER. The spectrum of surgical jaundice in
infancy. J Pediatr Surg. 2003;38:1471-9.
16. Superina R, Magee JC, Brandt ML, Healey PJ,
Tiao G, Ryckman F, et al. The anatomic pattern of biliary
atresia identified at time of Kasai hepatoportoenterostomy and early
postoperative clearance of jaundice are significant predictors of
transplant-free survival. Ann Surg. 2011;254:577-85.
17. Markowitz J, Daum F, Kahn EI, Schneider KM,
So HB, Altman RP, et al. Arteriohepatic dysplasia. I.
Pitfalls in diagnosis and management. Hepatology. 1983;3:74-6.
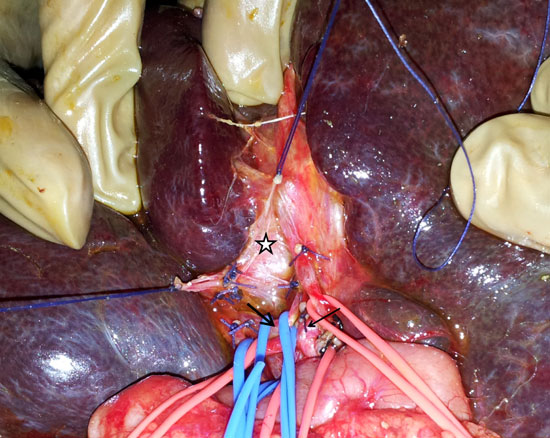
18. Kasai M, Suzuki S. A new operation for non
correctable biliary atresia: Hepatic portoenterostomy. Shijitsu.
1959;13:733-9.
19. Davenport M, De Ville de Goyet J, Stringer
MD, Mieli-Vergani G, Kelly DA, McClean P, et al. Seamless
management of biliary atresia in England and Wales (1999-2002).
Lancet. 2004;363:1354-7.
20. Davenport M, Ong E, Sharif K, Alizai N,
McClean P, Hadzic N, et al. Biliary atresia in England and
Wales: Results of centralization and new benchmark. J Pediatr Surg.
2011;46:1689-94.
21. Chardot C, Carton M, Spire-Bendelac N, Le
Pommelet C, Golmard JL, Auvert B. Prognosis of biliary atresia in
the era of liver transplantation: French national study from 1986 to
1996. Hepatology. 1999;30:606-11.
22. Karrer FM, Price MR, Bensard DD, Sokol RJ,
Narkewicz MR, Smith DJ, et al. Long-term results with the
Kasai operation for biliary atresia. Arch Surg. 1996;131:493-6.
23. Okazaki T, Kobayashi H, Yamataka A, Lane GJ,
Miyano T. Long-term postsurgical outcome of biliary atresia. J
Pediatr Surg. 1999;34:312-5.
24. Shinkai M, Ohhama Y, Take H, Kitagawa N, Kudo
H, Mochizuki K, et al. Long-term outcome of children with
biliary atresia who were not transplanted after the Kasai operation:
>20-year experience at a children’s hospital. J Pediatr
Gastroenterol Nutr. 2009;48:443-50.
25. van Heurn LW, Saing H, Tam PK.
Portoenterostomy for biliary atresia: Long-term survival and
prognosis after esophageal variceal bleeding. J Pediatr Surg.
2004;39:6-9.
26. Hung PY, Chen CC, Chen WJ, Lai HS, Hsu WM,
Lee PH, et al. Long-term prognosis of patients with biliary
atresia: A 25 year summary. J Pediatr Gastroenterol Nutr.
2006;42:190-5.
27. Sanghai SR, Shah I, Bhatnagar S, Murthy A.
Incidence and prognostic factors associated with biliary atresia in
Western India. Ann Hepatol. 2009;8:120-2.
28. Mohanty MK, Gupta SD, Bhatnagar V. Surgical
outcome in relation to duct size at the porta hepatis and the use of
cholagogues in patients with biliary atresia. Trop Gastroenterol.
2010;31:184-9.
29. Roy P, Chatterjee U, Ganguli M, Banerjee S,
Chatterjee SK, Basu AK. A histopathological study of liver and
biliary remnants with clinical outcome in cases of extrahepatic
biliary atresia. Indian J Pathol Microbiol. 2010;53:101-5.
30. Gupta L, Gupta SD, Bhatnagar V. Extrahepatic
biliary atresia: Correlation of histopathology and liver function
tests with surgical outcomes. J Indian Assoc Pediatr Surg.
2012;17:147-52.
31. Shimadera S, Iwai N, Deguchi E, Kimura O, Ono
S, Fumino S, et al. Significance of ductal plate malformation
in the postoperative clinical course of biliary atresia. J Pediatr
Surg. 2008;43:304-7.
32. Kawarasaki H, Itoh M, Mizuta K, Tanaka H,
Makuuchi M. Further observations on cystic dilatation of the
intrahepatic biliary system in biliary atresia after hepatic
portoenterostomy: report on 10 cases. Tohoku J Exp Med.
1997;181:175-83.
33. Inoue Y, Kato Y, Tamura T, Kobayashi H,
Ichikawa S, Lane GJ, et al. Prognostic implications of bile
lakes after surgery for biliary atresia. J Pediatr Surg.
2008;43:2165-8.
34. Pacheco MC, Campbell KM, Bove KE. Ductal
plate malformation-like arrays in early explants after a Kasai
procedure are independent of splenic malformation complex (heterotaxy).
Pediatr Dev Pathol. 2009;12:355-60.
35. Baerg J, Zuppan C, Klooster M. Biliary
atresia—a fifteen-year review of clinical and pathologic factors
associated with liver transplantation. J Pediatr Surg.
2004;39:800-3.
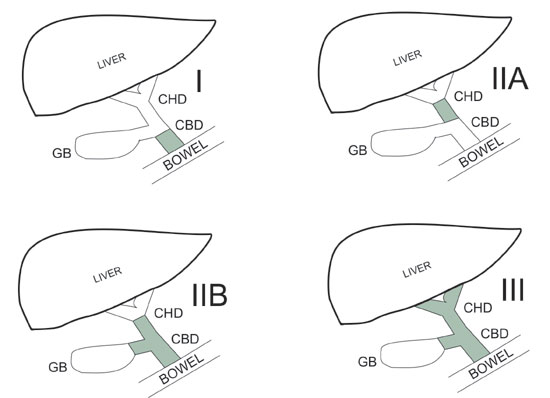
36. Tan CE, Davenport M, Driver M, Howard ER.
Does the morphology of the extrahepatic biliary remnants in biliary
atresia influence survival? A review of 205 cases. J Pediatr Surg.
1994;29:1459-64.
37. Langenburg SE, Poulik J, Goretsky M, Klein
AA, Klein MD. Bile duct size does not predict success of
portoenterostomy for biliary atresia. J Pediatr Surg.
2000;35:1006-7.
38. Wildhaber BE, Coran AG, Drongowski RA,
Hirschl RB, Geiger JD, Lelli JL, et al. The Kasai
portoenterostomy for biliary atresia: A review of a 27-year
experience with 81 patients. J Pediatr Surg. 2003;38:1480-5.
39. Grieve A, Makin E, Davenport M. Aspartate
aminotransferase-to-platelet ratio index (APRi) in infants with
biliary atresia: Prognostic value at presentation. J Pediatr Surg.
2013;48:789-95.
40. Suominen JS, Lampela H, Heikkila P, Lohi J,
Jalanko H, Pakarinen MP. APRi predicts native liver survival by
reflecting portal fibrogenesis and hepatic neovascularization at the
time of portoenterostomy in biliary atresia. J Pediatr Surg. 2014. pii:
S0022-3468(14)00792-1. doi:10.1016/j.jpedsurg.2014.11.046.
41. Nio M, Sasaki H, Wada M, Kazama T, Nishi K,
Tanaka H. Impact of age at Kasai operation on short- and long-term
outcomes of type III biliary atresia at a single institution. J
Pediatr Surg. 2010;45:2361-3.
42. Davenport M, Puricelli V, Farrant P, Hadzic
N, Mieli-Vergani G, Portmann B, et al. The outcome of the
older (> or =100 days) infant with biliary atresia. J Pediatr Surg.
2004;39:575-81.
43. Chen G, Zheng S, Sun S, Xiao X, Ma Y, Shen W,
et al. Early surgical outcomes and pathological scoring
values of older infants (>/= 90 d old) with biliary atresia. J
Pediatr Surg. 2012;47:2184-8.
44. Chardot C, Carton M, Spire-Bendelac N, Le
Pommelet C, Golmard J, Reding R, et al. Is the Kasai
operation still indicated in children older than 3 months diagnosed
with biliary atresia? J Pediatr. 2001;138:224-8.
45. Wildhaber BE. Biliary atresia: 50 years after
the first kasai. ISRN Surg. 2012;2012:132089.
46. Davenport M, Kerkar N, Mieli-Vergani G, Mowat
AP, Howard ER. Biliary atresia: The King’s College Hospital
experience (1974-1995). J Pediatr Surg. 1997;32:479-85.
47. Ohkohchi N, Chiba T, Ohi R, Mori S. Long-term
follow-up study of patients with cholangitis after successful Kasai
operation in biliary atresia: Selection of recipients for liver
transplantation. J Pediatr Gastroenterol Nutr. 1989;9: 416-20.
48. Bezerra JA, Spino C, Magee JC, Shneider BL,
Rosenthal P, Wang KS, et al. Use of corticosteroids after
hepatoportoenterostomy for bile drainage in infants with biliary
atresia: The START randomized clinical trial. JAMA. 2014;311:1750-9.
49. Davenport M, Grieve A. Maximizing Kasai
portoenterostomy in the treatment of biliary atresia: medical and
surgical options. S Afr Med J. 2012;102: 865-7.
50. Srinivasan P, Bowles MJ, Muiesan P, Heaton
ND, Rela M. Living related liver transplantation in biliary atresia
with absent inferior vena cava. Liver Transpl. 2001;7:376-7.
51. Dulundu E, Sugawara Y, Kaneko J, Kishi Y,
Akamatsu N, Matsui Y, et al. Short hepatic vein
reconstruction in biliary atresia patients with absent inferior vena
cava. Clin Transplant. 2007;21:13-7.
52. Falchetti D, de Carvalho FB, Clapuyt P, de
Ville de Goyet J, de Hemptinne B, Claus D, et al. Liver
transplantation in children with biliary atresia and polysplenia
syndrome. J Pediatr Surg. 1991;26:528-31.
53. Vincenzi R, Seda-Neto J, Fonseca EA, Ketzer
BM, Benavides M, Candido HL, et al. Technical aspects and
outcomes of living donor liver transplantation for pediatric
patients with situs inversus. Liver Transpl. 2013;19:431-6.
54. Kasahara M, Sakamoto S, Shigeta T, Uchida H,
Hamano I, Kanazawa H, et al. Reducing the thickness of left
lateral segment grafts in neonatal living donor liver
transplantation. Liver Transpl. 2013;19:226-8.
55. Kasahara M, Fukuda A, Yokoyama S, Sato S,
Tanaka H, Kuroda T, et al. Living donor liver transplantation
with hyperreduced left lateral segments. J Pediatr Surg.
2008;43:1575-8.
56. Vilca Melendez H, Vougas V, Muiesan P,
Andreani P, Mieli-Vergani G, Rela M, et al. Bowel perforation
after paediatric orthotopic liver transplantation. Transpl Int.
1998;11:301-4.
57. Tzakis AG, Kirkegaard P, Pinna AD, Jovine E,
Misiakos EP, Maziotti A, et al. Liver transplantation with
cavoportal hemitransposition in the presence of diffuse portal vein
thrombosis. Transplantation. 1998;65:619-24.
58. Lipshutz GS, Patel S, Hiatt JR, Yersiz H,
Farmer DG, McDiarmid SV, et al. Portocaval hemitransposition
in pediatric liver transplant recipients: A single-center
experience. Liver Transpl. 2006;12:1097-103.
59. Chardot C, Herrera JM, Debray D, Branchereau
S, De Dreuzy O, Devictor D, et al. Portal vein complications
after liver transplantation for biliary atresia. Liver Transpl Surg.
1997;3:351-8.
60. Utterson EC, Shepherd RW, Sokol RJ, Bucuvalas
J, Magee JC, McDiarmid SV, et al. Biliary atresia: Clinical
profiles, risk factors, and outcomes of 755 patients listed for
liver transplantation. J Pediatr. 2005;147:180-5.
61. Fouquet V, Alves A, Branchereau S, Grabar S,
Debray D, Jacquemin E, et al. Long-term outcome of pediatric
liver transplantation for biliary atresia: A 10-year follow-up in a
single center. Liver Transpl. 2005;11:152-60.
62. Diem HV, Evrard V, Vinh HT, Sokal EM, Janssen
M, Otte JB, et al. Pediatric liver transplantation for
biliary atresia: Results of primary grafts in 328 recipients.
Transplantation. 2003;75:1692-7.
63. Barshes NR, Lee TC, Balkrishnan R, Karpen SJ,
Carter BA, Goss JA. Orthotopic liver transplantation for biliary
atresia: The U.S. experience. Liver Transpl. 2005;11:1193-200.
64. Rao S, D’Cruz AL, Aggarwal R, Chandrashekar
S, Chetan G, Gopalakrishnan G, et al. Pediatric liver
transplantation: A report from a pediatric surgical unit. J Indian
Assoc Pediatr Surg. 2011;16:2-7.
65. Malhotra S, Sibal A, Bhatia V, Kapoor A,
Gopalan S, Seth S, et al. Living related liver
transplantation for biliary atresia in the last 5 years: Experience
from the first liver transplant program in India. Indian J Pediatr.
2015. doi:10.1007/s12098-014-1687-x
66. Group SR. Studies of pediatric liver
transplantation (SPLIT): Year 2000 outcomes. Transplantation.
2001;72:463-76.
67. Sandler AD, Azarow KS, Superina RA. The
impact of a previous Kasai procedure on liver transplantation for
biliary atresia. J Pediatr Surg. 1997;32:416-9.
68. Ng VL, Alonso EM, Bucuvalas JC, Cohen G, Limbers CA, Varni JW,
et al. Health status of children alive 10 years after pediatric
liver transplantation performed in the US and Canada: Report of the
studies of pediatric liver transplantation experience. J Pediatr.
2012;160:820-6.