|
|
|
Indian Pediatr 2012;49: 811 -818 |
 |
Immune Thrombocytopenic Purpura: Historical
Perspective, Current Status, Recent Advances and Future
Directions
|
|
P Anoop
From Department of Pediatric Hemato-Oncology, Great
Ormond Street Hospital for Children, London, United Kingdom.
Correspondence to: Dr P Anoop, Department of Pediatric
Hemato-Oncology, Apollo Hospital, Bannerghatta Road,
Bangalore 560 076, India.
Email: docanoop@yahoo.com
|
|
Immune thrombocytopenic purpura (ITP) has witnessed many changes and
updates over the past decade. The definitions of disease subtypes,
course and response to treatment have all been standardized recently.
Consequent to the lack of an international consensus management
guideline, wide variations exist in treatment practice. This is now
being addressed to an extent by the much awaited ITP International
Working Group 2010 recommendations. The pathophysiologic mechanisms have
been unfolded at cellular, molecular and humoral levels. As a result,
many recent advances have taken place in the management of this
disorder. This review revisits the history of evolution of ITP,
summarizes the current recommendations for management and lists the
recent advances and future prospects in this field.
Key words: Childhood, Immune thrombocytopenic
purpura.
|
|
I
mmune thrombocytopenic purpura
(ITP) is variably known as autoimmune thrombocytopenic purpura or immune
thrombocytopenia. It is no longer considered ‘idiopathic’. Although the
recent international consensus meeting suggested the use of the phrase
‘immune thrombocytopenia’ which includes both primary and secondary
subtypes, the familiar terminology of ITP will be adhered to here [1].
ITP has had a long and eventful journey with respect to the
understanding of its pathophysiology, diagnostic features and emerging
treatment modalities. This review revisits the evolution of this
disorder and provides an update on the current definitions, treatment
guidelines, recent advances in the field and likely future developments.
Population-based studies have shown that ITP has an
incidence of up to 6.4 per 100000 children and 3.3 per 100000 adults per
year [2]. The disorder is believed to differ biologically between them,
although similarities exist. The diagnosis and management of a typical
presentation of childhood ITP is usually not difficult. However,
thrombocytopenia secondary to other causes can often confound the
picture at presentation. Likewise, children who develop chronic
refractory thrombocytopenia can be challenging to treat.
Historical Overview
The term purpura originates from the Greek word
porphyra, a Mediterranean mollusc which yields a purple dye. The first
classical description of ITP was in 1735, by the German poet Paul
Werlhof (Fig. 1a). He referred to a young lady,
"without manifest cause, who bled from her nose and mouth and vomited
very thick, extremely black blood". He could identify that "about the
neck and on the arms, spots partly black, partly violaceous or purple
appeared" [3]. As a tribute to this description, ITP is now eponymously
known as Werlhof’s disease.
The existence of platelets remained elusive until
1874, when the Canadian physician William Osler (Fig. 1b)
drew and described ‘pale granular masses’ that circulated in the blood
[4]. He observed that they agglutinated when removed from circulation,
but believed these to be microorganisms. In 1881, the Italian
pathologist Giulio Bizzozero (Fig. 1c) delineated
the fundamental role of platelets in hemostasis. In 1889, George Hayem
proved the link between purpura and thrombocytopenia by physically
performing a platelet count on a patient [5].
 |
|
Fig.1 Key figures associated with the
evolution of ITP: (a) Werlhof, (b) Osler, (c) Bizzozero, (d)
Kaznelson. These images are available in the public domain.
|
The initial popular hypothesis was that ITP resulted
from limited production of platelets. This was challenged by a
Czechoslovakian medical student Paul Kaznelson (Fig. 1d),
who implicated excessive destruction of platelets by the spleen. In
1916, he persuaded his professor Schloffer to splenectomize a woman with
long standing ITP. The preoperative platelet count of 2 rose to 500 × 10 9/L
following splenectomy [5]. The famous Harrington experiment conducted in
Missouri in 1951 proved beyond doubt that a humoral factor in the plasma
was responsible for platelet destruction. William Harrington, a fellow
in hematology at Barnes-Jewish hospital, organized an exchange
transfusion of 0.5 L of whole blood between himself and a woman with
chronic purpura, whose blood group was the same as his [6]. Prior to the
exchange, Harrington and the patient had platelet counts of 250 and 5 ×
109/L
respectively. After exchange, the patient’s count remained unaltered at
6 × 109/L.
Harrington’s own count had dropped to 10 × 109/L
but normalized within a week.
Once the etiopathology of ITP was elucidated, various
treatment options evolved over time. The British dermatologist Robert
Willan had initially ‘prescribed’ "open fresh air, moderate exercise, a
generous diet and the free use of wine" [5]. Following Kaznelson’s
theory, splenectomy became the definitive treatment and remained so for
many decades. Other modalities tried included splenic irradiation,
injection of snake venom and irradiation by mercury vapour lamps. The
American hematologist Maxwell Wintrobe introduced immuno-suppressive
therapy with corticosteroids in 1951 [7]. The realization of the role of
Fc receptors on splenic macrophages led to the first successful use of
intravenous immunoglobulin (IVIG) by Paul Imbach in Switzerland in 1981
[8]. Two years later, James Bussel and his colleagues from New York
introduced anti-D therapy [9]. Advances in molecular biology and
targeted therapy then paved the way for the use of monoclonal antibodies
and thrombopoietin (TPO) agonists, described later [10-12].
Diagnostic Considerations
ITP is a diagnosis of exclusion. The combination of
patient history and clinical examination is by far the most important
diagnostic tool. At presentation, the clinician should consider the
various causes for secondary thrombocytopenia (Table I).
Atypical features which should prompt the pediatrician to think away
from the diagnosis of ITP include a very young age (relatively rare
under 2 years), family history of thrombocytopenia or bleeding,
significant lymphadenopathy, hepato-splenomegaly (mild splenomegaly is
described in young children with ITP), anemia disproportionate to the
degree of bleeding, leukocytosis or leukopenia, deranged clotting screen
and a systemically unwell child. Liver and renal function tests,
screening for viral infections such as HBV, HCV or HIV and an autoimmune
profile may help in selected cases. The vast majority of children who
clinically fit with a diagnosis of ITP do not require a bone marrow (BM)
examination [13]. Other expensive tests such as immunoglobulin levels,
monoclonal antibody-specific immobilization of platelet antigens
(MAIPA), platelet survival studies and reticulated platelets are not
recommended in typical presentations of ITP. Common variable immune
deficiency (CVID) is an important differential diagnosis from an Indian
perspective, as children can often present with isolated
thrombocytopenia.
TABLE I Causes of Thrombocytopenia in Children
|
Increased destruction |
Impaired
production |
|
Immune |
Aplasia / dysplasia |
|
Autoimmune: ITP, SLE, APLS, ES, CVID |
Chromosomal abnormalities, Thrombocytopenia absent radius,
Congenital amegakaryocytic thrombocytopenia, Fanconi anemia,
Dyskeratosis congenita, Myelodysplastic syndrome,
Myelofibrosis, Pearson syndrome |
|
Alloimmune: NAIT, PTP |
|
|
Drugs: Heparin induced thrombocytopenia |
|
|
Non-immune |
Marrow replacement |
|
Neonatal: TORCH infections, maternal disorders, birth |
Leukemia, solid tumours, storage disorders, histiocytosis,
|
asphyxia, Respiratory distress syndrome, Necrotizing
enterocolitis
Any age: Infections*, Disseminated intravascular coagulation,
Thrombotic thrombocytopenic purpura, Hemolytic uremic
syndrome, drugs, Kasabach Merritt syndrome, hypersplenism |
osteopetrosis |
|
Hereditary |
|
Bernard Soulier syndrome, Wiskott Aldrich syndrome,
May-Hegglin, von Willebrand disease 2b |
|
|
ITP, immune thrombocytopenic purpura; SLE, systemic lupus
erythematosus; APLS, anti-phospholipid syndrome; ES, Evans
syndrome; CVID, common variable immune deficiency; NAIT,
neonatal alloimmune thrombocytopenia; PTP, post-transfusion
purpura; * Hepatitis viruses B and C, human immunodeficiency
virus and H.pylori have proven associations with
thrombocytopenia. |
Complete blood count and blood film examination by a
hematopathologist are usually sufficient to support a clinical suspicion
of classical ITP. Morphological features include large platelets on a
peripheral blood smear (Fig. 2a, 2b) and an
adequate or increased number of megakaryocytes in the BM (Fig.
2c, 2d). In spite of the abundance of megakaryocytes,
they produce suboptimal number of platelets [14]. The most important
information to be established from blood and marrow examination is the
presence of normal erythrocytes, leukocytes and their precursors,
thereby excluding other hematological and infiltrative causes (Table
I).
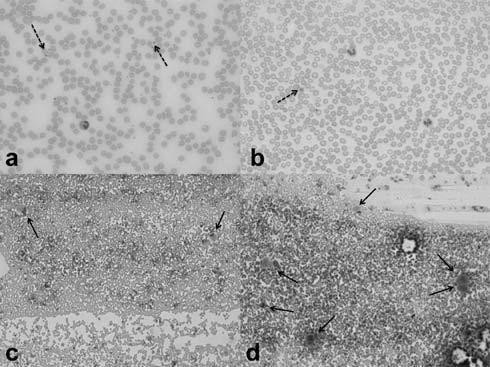 |
|
Fig. 2 Platelets and megakaryocytes.
(a) Peripheral smear showing normal platelets (dashed arrows)
(b) Peripheral smear showing thrombocytopenia with a large
platelet (dashed arrow) in ITP (c) BM with normal number of
megakaryocytes (arrows) (d) BM aspirate showing increased
megakaryocytes (arrows) in ITP [May-Grunwald-Giemsa stain; a and
b, original magnification x 40; c and d, original magnification
x 10].
|
In October 2007, the International Working Group on
ITP revised the terminologies (Table II) [1]. The
previously popular term ‘acute ITP’ was withdrawn and the cut-off
duration for designation as ‘chronic ITP’ was changed from 6 to 12
months. This was because a considerable proportion of children and
adults were noted to achieve remission between the 6-12 month period
from diagnosis. ‘Newly diagnosed ITP’ and ‘persistent ITP’ are now the
recommended terminologies to refer to patients within 3 months and
between 3-12 months from diagnosis respectively. Chronic ITP occurs in
about 20% of children, with a higher risk if >10 years and/or platelet
count ≥20 × 10 9/L
at presentation [15]. About 37-50% of children with chronic ITP achieve
remission within 4 years of diagnosis [15,16].
TABLE II Standardized Terminology Related to ITP (Adapted From the Recommendations of the Vicenza Conference, October 2007)
| Terminology |
Current
definition |
| Platelet threshold |
<100 × 109/L
(previously defined as <150 × 109/L) |
| Primary ITP |
Absence of secondary
causes to account for thrombocytopenia (diagnosis of exclusion) |
| Secondary ITP |
Immune thrombocytopenia
due to disease or drug exposure |
| Severe ITP |
Bleeding needing
treatment regardless of platelet count |
| Newly diagnosed ITP |
From diagnosis to 3
months (previously known as acute ITP until 6 months from
diagnosis) |
| Persistent ITP |
3-12 months after
diagnosis |
| Chronic ITP |
>12 months after
diagnosis (previously defined as >6 months after diagnosis) |
| Steroid dependent ITP |
Need for continuation
of steroids for at least 2 months to maintain platelet count
≥30 × 109/L and avoid
bleeds |
| Complete response (CR) |
Platelet count
≥100 × 109/L + no
bleeding |
| Response (R) |
Platelet count
≥30 × 109/L + at least
2-fold increase from baseline platelet count + no bleeding |
| No response (NR) |
Platelet count <30 × 109/L
(or) less than 2-fold increase from baseline platelet count (or)
bleeding |
| Refractory ITP |
Failure to achieve at
least R (or) loss of R after splenectomy + need for treatment to
control bleeding |
Treatment Considerations
Following the 2010 international consensus statement,
the American Society of Hematology (ASH) updated their ITP management
guidelines in 2011, providing evidence-based grades of recommendations (Table
III) [17,18].
TABLE III Grades of Recommendations for the Management of ITP
|
Clinical situation |
Recommendation |
Grade of evidence |
| New
presentation of suspected ITP |
BM
examination is not required if typical features |
Grade 1B |
|
are present |
|
| No bleeding
or cutaneous bleeds only |
Observation
alone |
Grade 1B |
| First line
medications |
IVIG |
Grade 1B |
|
Corticosteroids |
Grade 1B |
|
Anti-D |
Grade 2B |
| Non-response
to first line medications |
Rituximab |
Grade 2C |
| and recurrent
mucosal bleeds |
Splenectomy |
Grade 1B |
| Timing of
splenectomy |
If severe
persistent thrombocytopenia |
Grade 2C |
|
with mucosal
bleeds for at least 12 |
|
|
months and
failure of 2nd line medications |
|
| Routine
immunization |
All
vaccinations including MMR to be given |
Grade 1B |
|
ITP, immune thrombocytopenic purpura; BM, bone marrow; IVIG,
intravenous immunoglobulin; MMR, measles mumps rubella. |
The majority of children achieve spontaneous
remission and do not suffer major bleeding complications despite a
platelet count <10 × 10 9/L.
The expectant ‘watch and wait’ policy of management is recommended for
such patients. In the absence of ‘wet bleeding’, the child does not
require hospitalization. The frequency of follow up blood counts should
be limited to every 1-2 weeks initially and lesser thereafter, in order
to avoid unnecessary hospital visits and anxiety. The incidence of
intracranial hemorrhage (ICH) in children with ITP is 0.1-0.5% and
cannot be predicted precisely with confidence [19]. Parents should be
educated about the natural history of ITP and must be warned against the
use of non-steroidal anti-inflammatory drugs, intramuscular injections
and contact sports. Routine activities, schooling and vaccinations using
the subcutaneous route should be encouraged. In children with platelet
count ≥30 × 109/L,
intramuscular vaccination followed by firm pressure for 5 minutes is
generally considered safe.
The decision to treat should not be based solely on
the platelet count and must take into account the severity of bleeding
and associated risk factors. The available therapeutic options and
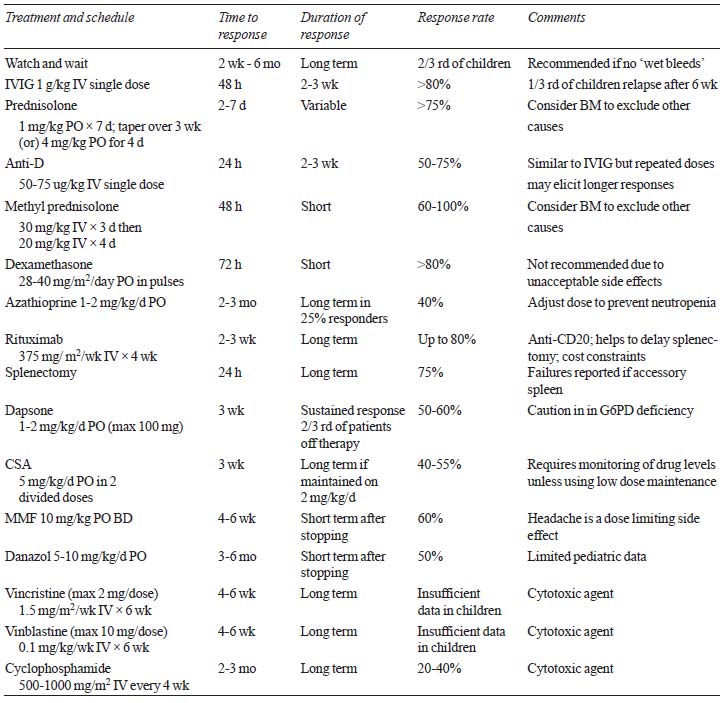
response rates are summarized in Table IV [15,17,20-24].
The recent ASH guideline regards IVIG, steroids and anti-D for children
with RhD positive blood groups as first line treatment options [18].
Immunomodulatory drugs for refractory cases include azathioprine,
cyclosporin A (CSA), mycophenolate mofetil (MMF), dapsone, danazol and
cytotoxic agents [20-24]. Four doses of rituximab at weekly intervals is
now emerging as an effective latter line of therapy in non-responders.
TABLE IV Therapeutic Options For Immune Thrombocytopenic Purpura
Splenectomy remains the definitive treatment for ITP
refractory to medical measures. Overwhelming post-splenectomy infection
(OPSI) is a significant complication, with maximal risk in children <5
years and especially <2 years of age [16]. Splenectomy should hence be
delayed beyond 6 years of age, preferably with severe thrombocytopenia
<10 × 10 9/L
and recurrent mucosal bleeds persistent for at least 12 months and
failure of second line medications [15,17,18,25]. Splenectomy has an
excellent and durable complete response (CR) rate of 75% [25,26].
Children should be vaccinated against pneumococcus, meningococcus and
H.influenzae
at least 2 weeks beforehand. The current recommendations for penicillin
prophylaxis is for until at least 5 years of age and for a minimum of 1
year after the procedure [16,27]. Many clinicians however, prefer to be
more conservative in this regard and recommend antibiotic prophylaxis
lifelong. Prior response to IVIG is associated with a higher success
rate after splenectomy, whereas non-response is not a predictor of
failure.
Platelet transfusions have no role, other than in
life threatening situations. For emergency treatment, a combination of
intravenous methyl prednisolone and IVIG/anti-D along with transfusion
of a supra-normal dose of platelets (up to 30 ml/kg) is recommended
[17]. Antifibrinolytics such as tranexamic acid 10-15 mg/kg
intravenously 6-hourly are useful to control bleeding. For elective
surgery, the desirable platelet count cut-off is dependent on the
bleeding risk. Thresholds of ≥30,
≥50 and ≥100
× 10 9/L
are suggested for low risk, high risk and neurosurgery, respectively
[19].
Recent Advances
Sophisticated immunological and molecular techniques
have facilitated a better understanding of the pathophysiology at
cellular and humoral levels. MAIPA has helped to demonstrate
autoantibodies against GpIIb/IIIa receptor on the platelet surface as
the basis for ITP [28,29]. Although not required for the clinical
work-up of suspected ITP, it is infrequently used for diagnosis in
patients with atypical presentation. Electron microscopic studies have
illustrated megakaryocyte changes including non-classic apoptosis,
cytoplasmic vacuolation, swelling of mitochondria and condensation of
nuclear chromatin [30]. Pathogenic roles of cytotoxic and regulatory
T-lymphocytes (Tregs) have also recently been recognized [31,32].
Reduced levels of TPO, the primary stimulator of platelet synthesis, is
now known to occur in patients with ITP [35]. TPO acts through its
receptor c-mpl to promote proliferation, differentiation and maturation
of megakaryocytes.
Therapeutic advances have paralleled the above
immunobiological breakthroughs, capitalizing on the unfolding of
causative mechanisms. The anti-CD20 monoclonal antibody rituximab is
emerging as the standard of care in children and adults with chronic
refractory ITP [10,17]. The initial excitement around others such as
anti-CD52 (alemtuzumab or campath-1H) and anti-CD40 ligand have not
translated into long term clinical remissions [11,34]. The TPO agonists
romiplostim and eltrombopag have recently successfully been through
safety and efficacy trials [12,35-37]. In an age-stratified, randomized,
placebo controlled study on 22 children with chronic ITP, Bussel, et
al. demonstrated that 88% achieved platelet count
≥50 × 10 9/L
after weekly subcutaneous
injections of romiplostim at a median dose of 5
mg/kg [35]. The phase III
randomized multicenter placebo controlled trial by Bussel, et al.
[36] on 114 adults with chronic ITP has reported a rise in platelet
count ³50
× 109/L
within 2 weeks in over half the
patients who received oral eltrombopag 50 mg daily. Despite these data,
it should be noted that TPO agonists are not yet approved for use in
children and hence are not currently recommended. Recombinant factor
VIIa has now been successfully used for hemostasis in patients with ITP
following life threatening bleeds [38].
Future Directions
Research so far has identified that ITP results from
both accelerated platelet destruction and defective production of
platelets by megakaryocytes. We also now know that both B and T
lymphocytes contribute to its pathogenesis, ie., autoimmunity of ITP has
both a humoral and a cellular basis. Dysregulation of the B-cell
survival pathway comprising of B-cell activating factor (BAFF), A
Proliferation Inducing Ligand (APRIL), B-cell maturation antigen (BCMA)
and their receptors has been proposed as an important component of
autoimmunity in ITP [39]. Therapeutic blockade of this pathway is a
promising treatment option for the future. Similar targeted
interventions against Tregs could also potentially reverse the
autoimmunity [31,32].
Newer monoclonal antibodies under investigation
currently include anti-Fc receptor (MDX-33), anti-Fc gRI
and anti-FcgRIII
(GMA-161) [40]. Experimentation with rozrolimupab, a symphobody against
the RhD antigen, is underway [41]. Another avenue being explored is
using an inhibitor of spleenz tyrosine kinase (Syk) [42]. It is also
hoped that romisplostim and eltrombopag, both now approved by FDA (USA)
and NICE (UK) for use in adults with refractory thrombocytopenia, will
accumulate more convincing safety data in children over the next decade.
Controversial Aspects in Diagnosis and Treatment
Age at presentation: Many pediatricians are
reluctant to diagnose ITP in young infants. Rarely, this disorder can
present in babies <6 months of age. Over a 2-year period at Great Ormond
Street Hospital (GOSH), we have followed up four babies aged under 6
months, in whom ITP was established as a diagnosis of thorough exclusion
of leukemia, bone marrow failure and immunodeficiency states. Maternal
platelet counts were normal in all cases; bone marrow showed increased
megakaryocytes and counts recovered either spontaneously or with IVIG/prednisolone.
Sandoval, et al. [43] reported ITP in 11 babies under 6 months of
age in their 15-year retrospective analysis. All patients responded
favourably to treatment, with a high rate of spontaneous remissions and
a low incidence of chronic ITP.
Need for marrow evaluation: There are reports of
missed leukemia and hemophagocytic lymphohistiocytosis (HLH) following
steroid therapy in patients wrongly diagnosed with ITP. Presence of
atypical features should make the clinician strongly consider a bone
marrow evaluation [13]. In resource-poor countries like India, it has
been argued that the accuracy of automated cell counters is not
uniformly reliable and hence a low threshold is required for assessing
the marrow [44].
Safe platelet count: There are wide discrepancies
in treatment practice across the world [45]. For years, the ASH had
recommended first line treatment with IVIG to achieve a ‘safe’ platelet
count ≥20 × 10 9/L
[26,45]. In contrast, the British Society for Haematology (BSH) favors
the watch and wait policy of management [45,46]. At GOSH, standard
practice is not to treat howsoever low the platelet count is, unless
there are additional risk factors or wet bleeds. It must be noted that
in their most recent 2011 updated guideline, ASH has also moved towards
the watch and wait policy [18].
Emerging role of rituximab: There is a general
consensus that splenectomy should be avoided as far as possible in
children. Rituximab now has good efficacy data with up to 80% response
rate in previously refractory patients. Currently the cost of this drug
precludes its use in many situations. With a potential reduction in cost
in future, rituximab may become the standard of care, thus obviating the
need for splenectomy in a significant proportion of children [25].
Safety concerns of TPO agonists: Despite the
efficacy of TPO agonists in refractory patients, concerns have been
raised on their long term safety [12]. Romiplostim has been associated
with venous and arterial thromboembolic events in adult patients.
Eltrombopag has caused hepatobiliary impairment in 10% of adults. Both
drugs have also been linked with bone marrow fibrosis.
Conclusions
There is now a reasonably clear understanding of the
pathophysiology of ITP. As a result, the repertoire of therapeutic
options has expanded. Clinicians should be aware of the strength of
evidence base for each modality of treatment. However, the most
important aspect in the management is to give adequate consideration to
alternate diagnoses at presentation. Atypical features are not uncommon
and should encourage the clinician to actively rule out secondary
thrombocytopenia. The management of refractory ITP patients with
recurrent bleeds, albeit rare, is quite challenging. Available second or
subsequent lines of therapy are to be used wisely in such situations,
weighing the benefits versus risks in each individual child. Referral to
a pediatric hematologist is recommended for children with unusual
presentations and resistant thrombocytopenia.
References
1. Rodeghiero F, Stasi R, Gernsheimer T, Michel M,
Provan D, Arnold DM, et al. Standardization of terminology,
definitions and outcome criteria in immune thrombocytopenic purpura of
adults and children: report from an International Working Group. Blood.
2009;113:2386-93.
2. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal
JB, George JN. The incidence of immune thrombocytopenic purpura in
children and adults: a critical review of published reports. Am J
Hematol. 2010;85:174-80.
3. Werlhof PG. Opera Omnia. Hanover: Helwing, 1775:
748. In: Major RH, editor. Classic Descriptions of Disease. 3rd
ed. Springfield, IL: CC Thomas, 1965.
4. Osler W. An account of certain organisms occurring
in the liquor sanguinis. Proc R Soc Med London. 1874;22: 391-9.
5. Stasi R, Newland AC. ITP: a historical
perspective. Br J Haematol. 2011;153:437-50.
6. Harrington WJ, Minnich V, Hollingsworth JW, Moore
CV. Demonstration of a thrombocytopenic factor in the blood of patients
with thrombocytopenic purpura. J Lab Clin Med. 1951;38:1-10.
7. Wintrobe MM, Cartwright GE, Palmer JG, Kuhns WJ,
Samuels LT. Effect of corticotrophin and cortisone on the blood in
various disorders in man. Arch Int Med. 1951;88:310-36.
8. Imbach P, d’Apuzzo V, Hirt A, Rossi E, Vest M,
Barandun S, et al. High-dose intravenous gamma globulin for
idiopathic thromboctyopenic purpura in childhood. Lancet.
1981;1:1228-31.
9. Bussel, JB, Graziano JN, Kimberly RP, Pahwa S,
Aledort LM. Intravenous anti-D treatment of immune thrombocytopenic
purpura: analysis of efficacy, toxicity, and mechanism of effect. Blood.
1991;77:1884-93.
10. Saleh MN, Gutheil J, Moore M, Bunch PW, Butler J,
Kunkel L, et al. A pilot study of the anti-CD20 monoclonal
antibody rituximab in patients with refractory immune thrombocytopenia.
Semin Oncol. 2000;27:99-103.
11. Willis F, Marsh JC, Bevan DH, Killick SB, Lucas
G, Griffiths R, et al. The effect of treatment with Campath-1H in
patients with autoimmune cytopenias. Br J Haematol.
2001;114:891-8.
12. Imbach P, Crowther M. Thrombopoietin receptor
agonists for primary immune thrombocytopenia. N Engl J Med.
2011;365:734-41.
13. Anoop P. Decision to perform bone marrow
aspiration in immune thrombocytopenic purpura must be based on evidence.
Pediatr Hematol Oncol. 2008;25:91-2.
14. Nugent D, McMillan R, Nichol JL, Slichter SJ.
Pathogenesis of chronic immune thrombocytopenia: increased platelet
destruction and/or decreased platelet production. Br J Haematol.
2009;146:585-96.
15. De Mattia D, Del Vecchio GC, Russo G, De Santis
A, Ramenghi U, Notarangelo L, et al; AIEOP-ITP Study Group.
Management of chronic childhood immune thrombocytopenic purpura: AIEOP
Consensus Guidelines. Acta Haematol. 2010;123:96-109.
16. Blanchette V. Childhood chronic immune
thrombo-cytopenic purpura (ITP). Blood Rev. 2002;16:23-6.
17. Provan D, Stasi R, Newland AC, Blanchette VS,
Bolton-Maggs P, Bussel JB, et al. International consensus report
on the investigation and management of primary immune thrombocytopenia.
Blood. 2010;115:168-86.
18. Neunert C, Lim W, Crowther M, Cohen A, Solberg L
Jr, Crowther MA. The American Society of Hematology 2011 evidence-based
practice guideline for immune thrombocytopenia. Blood.
2011;117:4190-207.
19. Imbach P, Kuhne T, Muller D, Berchtold W,
Zimmerman S, Elalfy M, et al. Childhood ITP: 12 months follow-up
data from the prospective registry I of the Intercontinental Childhood
ITP Study Group (ICIS). Pediatr Blood Cancer. 2006;46:351-6.
20. Hilgartner MW, Lanzkowsky P, Smith CH. The use of
azathioprine in refractory idiopathic thrombocytopenic purpura in
children. Acta Paediatr Scand. 1970;59:409-15.
21. Choudhary DR, Naithani R, Mahapatra M, Kumar R,
Mishra P, Saxena R. Efficacy of cyclosporine as a single agent therapy
in chronic idiopathic thrombocytopenic purpura. Haematologica.
2008;93:e61-e63.
22. Hou M, Peng J, Shi Y, Zhang C, Qin P, Zhao C,
et al. Mycophenolate mofetil (MMF) for the treatment of
steroid-resistant idiopathic thrombocytopenic purpura. Eur J Haematol.
2003;70:353-357.
23. Damodar S, Viswabandya A, George B, Mathews V,
Chandy M, Srivastava A. Dapsone for chronic idiopathic thrombocytopenic
purpura in children and adults: a report on 90 patients. Eur J Haematol.
2005;75:328-31.
24. Marwaha RK, Singh RP, Garewal G, Marwaha N,
Prakash D, Sarode R. Danazol therapy in immune thrombocytopenic purpura.
Pediatr Hematol Oncol. 1990;7:193-8.
25. Minkov M. Critical issues concerning splenectomy
for chronic idiopathic thrombocytopenic purpura in childhood. Pediatr
Blood Cancer. 2006;47:734-6.
26. George JN, Woolf SH, Raskob GE, Wasser JS,
Aledort LM, Ballem PJ, et al. Idiopathic thrombocytopenic purpura:
A practice guideline developed by explicit methods for the American
Society of Hematology. Blood. 1996;88:3-40.
27. Infectious Diseases and Immunization Committee,
Canadian Paediatric Society. Prevention and therapy of bacterial
infections for children with asplenia or hyposplenia. Paediatr
Child Health. 1999;4:417-31.
28. Kiefel V, Santoso S, Weisheit M, Mueller-Eckhardt
C. Monoclonal antibody–specific immobilization of platelet antigens
(MAIPA): a new tool for the identification of platelet-reactive
antibodies. Blood. 1987;70:1722-6.
29. Woods Jr VL, Oh EH, Mason D, McMillan R.
Autoantibodies against the platelet glycoprotein IIb/ IIIa complex in
patients with chronic ITP. Blood. 1984;63:368-75.
30. Houwerzijl EJ, Blom NR, van der Want JJ, Esselink
MT, Koornstra JJ, Smit JW, et al. Ultrastructural study shows
morphologic features of apoptosis and para-apoptosis in megakaryocytes
from patients with idiopathic thrombocytopenic purpura. Blood.
2004;103:500-6.
31. Olsson B, Andersson PO, Jernas M, Jacobsson S,
Carlsson B, Carlsson LM, et al. T-cell-mediated cytotoxicity
toward platelets in chronic idiopathic thrombocytopenic purpura. Nature
Med. 2003;9:1123-4.
32. Yu J, Heck S, Patel V, Levan J, Yu Y, Bussel JB,
et al. Defective circulating CD25 regulatory T cells in patients
with chronic immune thrombocytopenic purpura. Blood.
2008;112:1325-8.
33. Kuter DJ, Gernsheimer TB. Thrombopoietin and
platelet production in chronic immune thrombocytopenia. Hematol Oncol
Clin North Am. 2009;23:1193-211.
34. Patel VL, Schwartz J, Bussel JB. The effect of
anti-CD40 ligand in immune thrombocytopenic purpura. Br J Haematol.
2008;141:545-8.
35. Bussel JB, Buchanan GR, Nugent DJ, Gnarra DJ,
Bomgaars LR, Blanchette VS, et al. A randomized, double-blind
study of romiplostim to determine its safety and efficacy in children
with immune thrombocytopenia. Blood. 2011;118:28-36.
36. Bussel JB, Provan D, Shamsi T, Cheng G, Psaila B,
Kovaleva L, et al. Effect of eltrombopag on platelet counts and
bleeding during treatment of chronic idiopathic thrombocytopenic purpura:
a randomised, double-blind, placebo-controlled trial. Lancet.
2009;373:641-8.
37. Bussel JB, Cheng G, Saleh MN, Psaila B, Kovaleva
L, Meddeb, B et al. Eltrombopag for the treatment of chronic
idiopathic thrombocytopenic purpura. N Engl J Med.
2007;357:2237-47.
38. Salama A, Rieke M, Kiesewetter H, von Depka M.
Experiences with recombinant FVIIa in the emergency treatment of
patients with autoimmune thrombocytopenia: a review of the literature.
Ann Hematol. 2009;88:11-5.
39. Emmerich F, Bal G, Barakat A, Milz J, Muhle C,
Martinez-Gamboa L, et al. High-level serum B-cell activating
factor and promoter polymorphisms in patients with idiopathic
thrombocytopenic purpura. Br J Haematol. 2007;136: 309-14.
40. Li X, Hou M. Emerging drugs for idiopathic
thrombocytopenic purpura in adults. Expert Opin Emerg Drugs.
2008;13:237-54.
41. Stasi R. Rozrolimupab, symphobodies against
rhesus D, for the potential prevention of hemolytic disease of the
newborn and the treatment of idiopathic thrombocytopenic purpura. Curr
Opin Mol Ther. 2010;12:734-40.
42. Podolanczuk A, Lazarus AH, Crow AR, Grossbard E,
Bussel JB. Of mice and men: an open-label pilot study for treatment of
immune thrombocytopenic purpura by an inhibitor of Syk. Blood.
2009;113:3154-60.
43. Sandoval C, Visintainer P, Ozkaynak MF, Tugal O,
Jayabose S. Clinical features and treatment outcomes of 79 infants with
immune thrombocytopenic purpura. Pediatr Blood Cancer.
2004;42:109-12.
44. Naithani R, Kumar R, Mahapatra M, Agrawal N, Pati
HP, Choudhry VP. Is it safe to avoid bone marrow examination in
suspected ITP? Pediatr Hematol Oncol. 2007;24:205-7.
45. Anoop P. Variations in the treatment threshold
for immune thrombocytopenic purpura. Pediatr Blood Cancer.
2009;52:429-31.
46. British Committee for Standards in Haematology
General Haematology Task Force. Guidelines for the investigation and
management of idiopathic thrombocytopenic purpura in adults, children
and in pregnancy. Br J Haematol. 2003;120:574-96.
|
|
|
 |
|