|
|
Case Reports Indian Pediatrics 2002; 39:967-969 |
||
|
Von Voss-Cherstvoy Syndrome |
||
|
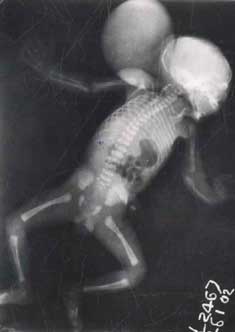
Von Voss-Cherstvoy syndrome or DK Phocomelia syndrome is a rare, perinatally lethal, unusual complex of multiple congenital anomalies, characterized by phocomelia especially radial ray defects, hypo- megakaryocytic thrombocytopenia, occipital encephalocoele, and urogenital abnormalities. We report a proposita with these characteristic anomalies and other associations. No case has been reported from India so far, to the best of our knowledge. Case Report A 20-year-old unbooked primigravida mother with severe oligohydramnios, delivered a 30-week male child weighing 1530 grams at Kasturba Hospital, M.G.I.M.S, Sevagram. The infant had multiple congenital anomalies, an Apgar of 1,3,3 at 1,5 and 10 minutes respectively and succumbed after two and half hours of life. There was no history of consanguinity in the parents, exposure to known teratogens or a family history of malformations. Mother did not receive folic acid during the periconceptional period. On clinical examination, there was a large posterior midline occipital encephalocele measuring 9 × 6.5 × 5.5 cm. Forearms were shortened and radially curved with-radial deviation of the hands, more on the left side. The thumbs were present on both sides. Other abnormal features included asymmetrical face, medial epicanthal folds, low set misshapen ears, compressed nose, and receded chin (Potter facies). The palate was high arched and uvula was bifid. Tongue was hypoplastic and there were natal teeth. Spine and lower limbs appeared normal. Umbilical cord and the placenta were normal. Platelet count was 56,000 per cubic mm. Rest of the hematological parameters were normal. Skeletal survey (Fig. 1) revealed a calvarial defect of the occipital bone posteriorly with a large encephalocele. The maxilla, mandible and the nasal bone were hypoplastic. There was bilateral pneumothorax. Upper limbs showed absent radius and radially curved hypoplastic ulna on the left side. The right radius was hypoplastic with a normal ulna. The radial rays were present on both the sides. Spine, pelvic and lower limb bones were normal. Cranial ultrasonography showed holoprocencephaly and absent corpus callosum. Sonography of the encephalocele revealed fluid and few unidentifiable brain structures.
Autopsy confirmed the above abnormal-ities. Both the lungs were hypoplastic. Thymus was absent. Heart showed an atrial and a large membranous ventricular septal defects with normal great vessels and their connections. Diaphragm was normal. Liver was enlarged and an accessory spleen was present in addition to a normal spleen. The urogenital anomalies included hypoplastic kidney and absent testis on the left side. The right kidney was enlarged due to hydronephrosis. Grossly dialated right hydroureter opened into a large urine filled bladder. There was bladder neck obstruction due to posterior uretheral valves. Histopathology of the organs was normal except for immature lungs and left kidney. Megakaryocytes were markedly reduced in the bone marrow and the liver (which showed extramedulary hemopoiesis). Karyotyping revealed 46, XY with G-banding. Discussion Von Voss et al.(1) and Cherstvoy et al.(2) first reported an unusual complex of multiple congenital anomalies including phocomelia, thrombocytopenia, occipital encephalocele, and urogenital abnormalities. It is a very rare condition and is referred to as von Voss-Cherstvoy syndrome, DK Phocomelia syndrome or Occipital encephalocele and MURCS (Mullerian ducts, Renal, Cervico-thoracic Spine) association(2,3,4) . Von Voss et al.(1) in 1979 described a girl aged 3 month with this unusual complex. They suggested it as a new syndrome because of the differences from all the variants of radial ray defects and thrombocytopenia. Cherstvoy et al.(2) in 1980 described an apparently identical case and proposed to call this new malformation syndrome as the ‘DK-phocomelia syndrome’, from the first letters of the children’s surnames–"D" for von Voss’s patient and "K" for their patient. The initial cases had occipital encephaloceles and phocomelia, but since milder cerebellar anomalies and arm findings limited to radial anomalies may be seen instead, Lubinsky et al.(3) suggested that the original designation of DK-phocomelia syndrome is inaccurate. They proposed to call this syndrome as "Von Voss-Cherstvoy syndrome". Etiology of Von Voss-Cherstvoy syndrome is not known. Both sexes are affected and parental age is not increased. Reports on two consanguineous families, one with recurrences, suggest autosomal recessive inheritance in at least some instances(3). All previously reported cases of Von Voss-Cherstvoy syndrome had a normal chromosomal constitution except a recently reported case in which the patient had normal lymphocyte chromosomes but was mosaic in fibroblasts for deletion of 13q 12(5). Pathogenesis on the malformations proposed is a defect in the organization of the paraxial mesoderm that gives rise to occipital, cervical, and thoracic somites and adjoining intermediate mesoderm. These structures contribute to the occipital bone, cervical spine, upper limbs, and urogenital system(4,6). Von Voss-Cherstvoy syndrome is a perinatally lethal syndrome but few cases reaching 8 years have been reported(2,5,7). Congenital anomalies affect upper limbs, face, brain, heart, lungs, urogenital and gastrointestinal systems, vertebrae and ribs, and includes thrombocytopenia(3). In our case the encephalocele was large posterior occipital in the midline as classically described by Von Voss et al. Phocomelia was limited to radial aplasia/hypoplasia with intact thumb. There was hypomegakaryocytic thrombocytopenia. Urogenital anomalies included hypoplastic left kidney, absent left testis and posterior uretheral valves. Apart from constellation of these features, our patient also had high arched palate, bifid uvula, hypoplastic tongue, natal teeth, thymic aplasia, atrial and ventricular septal defect. Absent thymus, natal teeth and posterior uretheral valves seen in our case, have not been described earlier. Contributors: SM diagnosed the condition and drafted the manuscript; he will act as guarantor. SK carried out the autopsy, hematological and histo-pathological investigations. SM and KV were involved in the management of the patient. KV and PC co-drafted and critically evaluated the paper. Funding: None. Competing interests: None stated.
| ||
|
References | ||
|
![]()