|
|
Brief Reports Indian Pediatrics 2001; 38: 1144-1148 |
||||
|
Outcome of Biliary Atresia from Chandigarh: Results of a Prospective Analysis |
||||
|
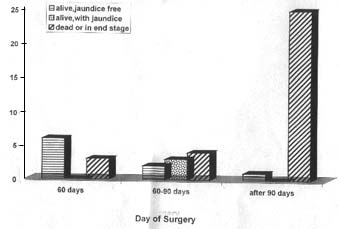
Biliary atresia is a disease of unknown etiology. If untreated it has a progressive course to end stage liver failure and death. With the advent of portoenterostomy and liver transplantation, children have a promise for long term survival. The results of treatment of biliary atresia in the developed nations have not been achieved in India. We undertook this prospective study to analyze the results of treatment of patients of biliary atresia in our population. Patients and Methods Forty four consecutive patients of surgical jaundice were included in the study. There were 34 males and 10 females. Medical causes for obstructive jaundice were investigated. The protocol included a clinical examination, examination of the stool color and liver function tests to confirm the presence of obstructive jaundice. A TORCH serology and HIDA scan were done. Ultrasound abdomen was routinely done to exclude the presence of choledochal cyst or a distended gallbladder. Liver biopsy was requested if the patient presented very late to avoid unnecessary surgery in frankly cirrhotic patients. All the clinical and operative data were prospectively entered. Preoperative correction of coagulopathy and hydration was routinely done. All babies with persistent white stools and conjugated hyperbilirubi-nemia after 8 weeks of life were subjected to an operative cholangiogram as soon as possible. Visualization of the extrahepatic biliary tree along with the intrahepatic branches was a definite proof to exclude biliary atresia and these patients had a liver biopsy and closure of the abdomen. Such patients were excluded from this analysis. In the absence of a patent extra hepatic biliary tree, a porto-enterostomy was done using a Roux en-Y porto enteric anastomosis with a 45 cm long loop of small bowel without an antireflux procedure. When there was no bile flow from the porta after excision of the biliary tract, further dissection of the porta between the tributaries of vessels and the portal vein was done. When the liver was frankly cirrhotic and the patient presented after 120 days, no porto-enterostomy was attempted. Postoperative steroids were used only if there was cessation of bile flow with fever. All patients were carefully followed up. Patients who did not turn up for follow up were contacted by telephone or letters. All excised tissue from the porta and the liver specimens were histopathologically analyzed. Results Nine babies were operated before 60 days of life and another nine between 61 days and 90 days of life (Fig. 1). Twenty six babies were operated beyond 90 days of life. Some of them were as old as four and a half months at the time of surgery. Six out of the nine patients operated before 60 days of life are alive and jaundice free. They have normal liver functions. Five of them have completed one year after surgery. One patient with associated duodenal atresia expired peri-operatively. Two patients initially had bile excretion which ceased after attacks of cholangitis eventually leading to death. Seven out of the nine patients operated between 60 to 90 days of life excreted bile after surgery. Two of these patients presented with increasing obstructive jaundice after one year. One of these patients was re-explored and the patient died perioperatively of hepatic insufficiency. Three children have signs of liver failure in the form of hypoalbuminemia with ascites. Only 2 children are anicteric and well. Even these children have elevated transaminases which suggests that these children may be having progressive disease. In these two groups, the incidence of cholangitis was 5 out of 18 (28%).
Fig. 1. Graph showing the outcome of patients (y axis) in relation to the day of surgery (X axis) The results of surgery after three months of age were disastrous. Successful bile excre-tion was established only in one child who was operated at ninety one days of life. He is one year postoperatively and is jaundice free. Six patients died in the first month after surgery, three perioperatively due to hepato-renal failure and three whose liver was cirrhotic and were not offered portoentero-stomy. In the remaining patients, successful bile excretion was not established and they dropped out of follow up. Associated anomalies were seen in 3 patients, namely G6PD deficiency, thalas-semia major and duodenal atresia with preportal duodenal vein. Of the 23 patho-logical specimens that were reanalyzed, 20 had both the specimens of porta and liver available. In 3 patients no luminal structure was discernable in the porta. In these patients there was no bile excretion. In two patients, a large luminal structure 700-800 microns was seen. In the rest, multiple channels of varying sizes were seen. These were lined by biliary epithelium, surrounded by scattered loose stroma, lymphoid follicles and extravasated bile pigments. In all these patients there was no correlation between the quantity of bile excretion and the size of the ducts. There was definite decrease in bile excretion after an attack of cholangitis. In the 23 specimens of liver, the overall morphology was similar. Three were frankly cirrhotic. In the rest there was varying degrees of portal fibrosis and bridging. In all the cases the degree of bile duct proliferation and extent of cholestasis was random and varied between moderate to severe. The number and size of the bile duct lumen seen in the porta seemed to have no direct influence on the extent of bile duct proliferation and cholestasis. The hepatocytes showed feathery degeneration or ballooning degeneration and there was moderate portal tract inflammation. Discussion Extrahepatic biliary atresia is a common life threatening disorder of infancy and affects at least 1 in 10,000 live births(1). Bile drainage can be restored by Kasai porto-enterostomy and surgery must be done before all the intrahepatic bile ducts leading to the porta hepatis are destroyed(2-5). Several studies confirm that surgery before 8 weeks of life is important to obtain effective bile drainage. Even in developed countries corrective surgery is delayed because of late referral(6). Long term survival data varied from 15.9% to 40.5% from various centers (2,7). Recent studies from Japan have suggested that though there is seemingly good prognosis in the post Kasai era in patients with good bile drainage, liver functions can progressively deteriorate around adolescence. Liver transplantation is necessary to obtain good survivals in biliary atresia(2). The published data from India discuss the clinical profile of patients operated and the complications but not the survivals(2-6). In our analysis, 13 out of 44 patients (29.5%) are alive after surgery, and 7(15%) have no signs of liver failure at the end of one year. Three of the nine children operated after 60 days of life have signs of liver failure at the end of one year. The five survivors operated before 60 days of life are faring better than those operated later. The results of surgery after 90 days of life are very poor. Though it has been recognized that early age at surgery is important in achieving good bile flow, analysis of median age of referral from over 100 institutes revealed a median age of 10 weeks at the time of surgery(6). In our center, 26 patients (60%) were referred for surgery after 90 days of life. Only 9 patients were operated before 60 days of life. Parents of all the babies who presented later had consulted a medical practitioner within the first 3 weeks of life for jaundice. Precious time was lost due to lack of awareness of urgency of the problem. Conjugated hyper-billirubinemia or presence of clay colored stools at the end of 2 weeks of life in a child is an urgent surgical problem and must be referred to an appropriate center. In our analysis of the liver histology there was no correlation between the degree of liver damage and the timing of surgery. The amount of bile drainage did not correlate with the size of the ducts at porta. Bile duct size at porta, degree of liver changes, amount of bile flow and attacks of cholangitis are known to affect the outcome after surgery for biliary atresia. The key factor that can make a difference in the results is early surgery. All the centers in India see less than 20% of their cases before 60 days of life(2-6). In Japan there were 9% long term survivors in the first decade of treatment, 20% in the second decade and 60% in the 3rd decade. The improvements in the 3rd decade was because of the availability of liver trans-plantation(2). Western countries have achieved over 85% graft survivals in a cadaver program whereas countries like Japan and Korea have developed live related programs for cultural reasons(8,9). We must encourage early referrals and early surgery. The upper age limit for surgery must not be too strict. Six out of the 80 survivors in a French study were over three months of age at the first operation(9). The outcome of surgery of children with biliary atresia has also been found to be related to the case load of the surgical center. In a study from U.K. and Ireland the mean age at surgery was 54 days and the outcome of surgery was better in centers having a higher case load(10). In conclusion, the technique of surgery and postoperative management of biliary atresia is now standardized in our country. The immediate postoperative outcome is comparable to the centers overseas. The hurdles lie with the impact on the long term outcome made by late referral and inherent problems with our medical infrastructure. Acknowledegments We wish to thank Ms Balpinder Kaur for her assistance in following up these patients. Contributors: KLN coordinated the study and prepared the paper; he will act as the guarantor for the manuscript. SKC, KLNR, RS and JK were the surgeons who participated in the study. They were involved in designing the study. They maintained their patient records and reviewed the manuscript critically. BRT was the pediatric gastroenterologist who helped in following the patients. KV was the pathologist who studied the specimens.
Funding:
None.
References
|
|
|