|
Reciprocal translocation
carriers are at the risk of having a mentally and physically abnormal
child because of "segmental aneusomy". The imbalance is due to
duplication or deletion of the chromosome segment involved in
segregation. Partial autosomal monosomies and trisomies, although
associated with congenital malformations, are known to be compatible
with life.
7q deletions have been reported in more than 30
cases as either an isolated deletion or in combination with other
chromosomal anomalies [1]. In most of the cases the associated
clinical features are highly variable, and are found to share a few
common features like microcephaly, broad nasal bridge, bulbous nasal
tip, auricular malformations, micro-gnathia and genital anomalies,
which delineate a distinct phenotype as ‘7q terminal deletion
syndrome’ [2]. Individuals with 7q monosomy are known to have
malformations like holoprosencephaly and mid structural defects [2]
and various grades of caudal deficiency sequence [3]. We herein
describe a male patient with partial 7q monosomy and partial trisomy
of 14q resulting from paternal t(7;14)(q33;q32.3).
Case Report
The patient presented to us as a 4-year-old male
born to unrelated parents. The couple had history of two previous
first trimester abortions. There was no history of antenatal intake
of drugs or any systemic illness in the mother. The patient’s birth
history was uneventful and birth weight was reported as average.
There was history of delayed milestones and at age of 4 years he was
not able to stand with support or feed himself. There was no speech
development and the patient was not able to obey commands. There was
no history of seizures or any other problems. On examination, the
patient had height of 91 cm (less than 5th centile), and head
circumference of 46.5 cm (less than 5th
centile).
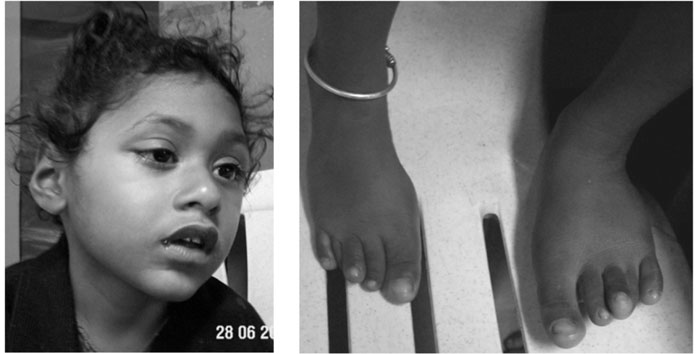
The dysmorphic features included
microcephaly, malformed low set ears, depressed nasal bridge, bulbous
nasal tip and over-riding of fingers and toes (Fig 1).
 |
|
Fig. 1 Facial features and overriding
toes in proband. |
His metabolic screening and MRI brain were normal.
Abdominal ultrasound for renal anomalies and echocardiography was
normal. Chromosomal analysis was performed on peripheral lymphocytes
following standard procedure. The karyotype at 550 band level, showed
an unbalanced chromosomal rearrangement (46, XY del [7] (q33-qter). A
total of 50 metaphases were analyzed and all showed abnormal
karyotype, thus ruling out mosaicism. Parental cytogenetic analysis
revealed a balanced reciprocal translocation between 7q and 14q in
father (46, XY, t(7;14)(q33;q32.3)). Maternal karyotype was normal.
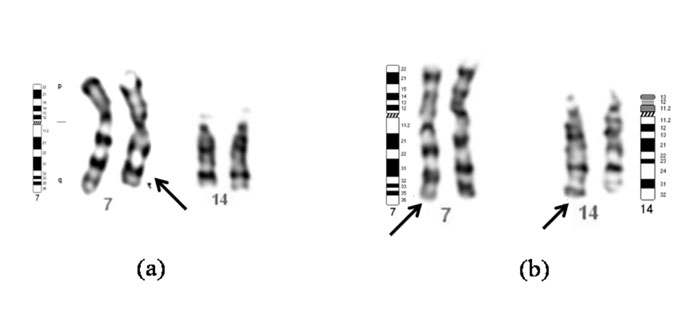
Consequently, the proband’s chromo-somal imbalance was interpreted as
partial monosomy 7q33-qter and partial trisomy 14q32.3-qter (46, XY,
der (7), t(7;14)(q33-q32.2)pat (Fig. 2).
 |
|
Fig. 2 (a) Partial karyotype
of the boy with partial monosomy 7q33 showing normal
chromosomes 7, 14 and a derivative chromosome 7 (arrow). (b)
Partial karyotype of the father with balanced
t(7;14)(q33;q32.3) showing normal and derivative (arrows)
chromosomes 7 and 14. |
Discussion
Our patient did not have premaxillary agenesis
single central incisor, holoprosencephaly, stigmata of caudal
deficiency sequence, or congenital cardiovascular, renal/urinary or
adrenal defect. Thus the 7q deletion in the index patient resulted in
a relatively mild phenotype as compared to the literature [3].
Our patient did not show significant cerebral
malformations, typical signs of holoprosencephaly, or relatively
severe manifestations of the syndrome including , caudal deficiency
sequence, congenital heart malformation as reported in most terminal
7q deletion cases [1-5,7,8]. The phenotype results from deletion of
various genes distal to the breakpoint on 7q like Sonic Hedgehog (SHH),
HLXB9 etc leading to haploinsufficiency of these genes. However our
patient is similar to cases with 7q deletion with a mild phenotype
[6,9,10]. This shows the wide phenotypic spectrum of cases with 7q
deletion.
Patients with partial trisomy 14q show significant
variability which may be due to the length of the 14q segment
involved, or the nature of the other chromosome segment involved in
the reciprocal translocation. The clinical features vary from severe
to mild dysmorphic features associated or not with chromosome
aberrations. The clinical features in our patient are consistent with
those described in 7q deletion syndrome. Although the patient also
had a small partial 14q trisomy, we cannot comment about the
significance of this in relation to patient’s phenotype. The typical
partial 14q trisomy phenotype was not seen in the proband probably
due to small size of partial trisomy or because this segment may
contain genes that are not subject to dosage effect.
The identification of balanced translocation in
the father helped us to counsel the family regarding risk of
unbalanced chromosomal rearrangements in fetuses in subsequent
pregnancies of the couple. The couple was counseled that there is
risk of chromosomal imbalance in all future pregnancies. They were
advised regarding availability of prenatal diagnosis and termination
of pregnancy.
This case report further emphasizes the importance
of doing karyotype in all cases of mental retardation, even if there
are few dysmorphic features. The testing of proband will help both in
counseling the parents regarding the cause of disease in their child
as well as prevention of recurrence of the abnormality in future
pregnancies.
Contributors: Both authors were involved in
data collection, analysis and drafting of the paper.
Funding: None.
Competing interests: None stated.
References
1. Frints SG, Schoenmakers EF, Smeets E, Petit P,
Fryns JP. De Novo 7q36 deletion: breakpoint analysis and types of
holoprosencephaly. Am J Med Genet. 1998;75:153-8.
2. Frints SG, Schrander-Stumper CT, Schoenmakers
EF, Engelen JJ, Reekers AB, Van Den Neucker AM, et al. Strong
variable clinical presentation in 3 patients with 7q terminal
deletion. Genet Couns. 1998;9:5-14.
3. Nowaczyk MJ, Huggins MJ, Tomkins DJ, Rossi E,
Ramsay JA, Woulfe J, et al. Holoprosencephaly, sacral
anomalies and situs ambiguious in an infant with partial monosomy 7q/trisomy
2p and SHH and XLXB9 haploinsufficiency. Clin Genet. 2000; 57:388-93.
4. Ahn JM, Koo DH, Kwon KW, Lee YK, Lee YH, Lee HH,
et al. Partial trisomy 2q(2q37.3®qter)
and monosomy 7q(7q34®qter)
due to paternal reciprocal translocation 2;7: a case report. J Korean
Med Sci. 2003;18:112-3.
5. Chen CP, Devriendt K, Lee CC, Chen WL, Wang W,
Wang TY. Prenatal diagnosis of partial trisomy 3p(3p23®pter)
and monosomy 7q(7q36®qer)
in a fetus with microcephaly, alobar holoprosencephaly and cyclopia.
Prenat Diagn. 1999;19:986-9.
6. Horn D, Tonnies H, Neitzel H, Wahl D, Hinkel
GK, Von Moers A, et al. Minimal clinical expression of the
holoprosencephaly spectrum and of Currarino syndrome due to different
cytogenetic rearrangements deleting the Sonic Hedgehog gene and the
HLXB9 gene at 7q36.3. Am J Med Genet. 2004;128:85-92.
7. Hehr U, Gross C, Diebold U, Wahl D, Beudt U,
Heidemann P, et al. Wide phenotypic variability in families
with holoprosencephaly and a sonic hedgehog mutation. Eur J Pediatr.
2004;163:347-52.
8. Su PH, Chen JY, Chen SJ, Tsao TF, Lai YJ.
Sacral dysge-nesis associated with terminal deletion of chromosome
7(q36®qter).
Pediatr Neonatol. 2008;49:189-92.
9. Lukusa T, Vermeesch JR, Fryns JP. De novo
deletion 7q36 resulting from distal 7q/8q translocation: Phenotypic
expression and comparison to the literature. Genet Couns.
2005;16:1-15.
10. Ginocchio VM, De Brasi D, Genesio R, Ciccone
R, Gimelli S, Fimiani F, et al. Sonic hedgehog deletion and
distal trisomy 3p in a patient microphthalamia and microcephaly,
lacking cerebral anomalies typical of holoprosencephaly. Eur J Med
Genet. 2008;51:658-65.
|


