|
|
Case Reports Indian Pediatrics 2007;44:371-374 |
||||
|
An Unusual Cause of Ascites: Hemophagocytic Lymphohistiocytosis |
||||
|
Mustafa Akcam From the Division of Pediatric Gastroenterology, Hepatology and Nutrition, Departments of Pediatrics and *Pathology, Akdeniz University School of Medicine, Antalya/Turkey.
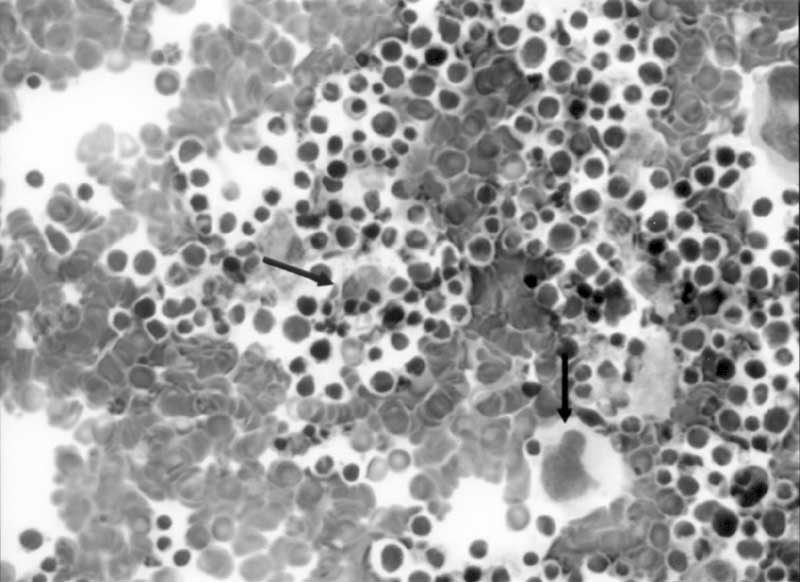
Abstract: Hemophagocytic lymphohistiocytosis is character-ized by fever, hepatosplenomegaly, cytopenia, hyper-triglyceridemia, hypofibrinogenemia, and hemophago-cytosis. Ascites is not mentioned as a symptom of hemophagocytic syndrome. We report a one month-old girl suffering from familial erythrophagocytic lympho-histiocytosis, who presented with ascites. Key words: Hemophagocytosis, ascites, childhood. Hemophagocytic lymphohistiocytosis (HLH) is a rare disease affecting infants and children. It has two forms with similar presentation: familial erythrophagocytic lymphohistiocytosis (FEL) and infection-associated hemophagocytic syndrome (IAHS)(1). The former has autosomal recessive inheritance with an incidence of 0.12 per 100.000 children(2). The hallmark of the disease is the accumulation of lymphohistiocytes in the reticuloendothelial system with features of hemophagocytosis. Although children with IAHS may present at an older age, children with FEL are always younger than 4 years(1-3). Ascites is rarely reported in patients with HLH in the literature(4,5). In this report, we are presenting an infant with FEL, who had an unusual presentation of ascites, and who was younger than the previously published ones. Case Report Thirty-five-days old female baby was referred to our hospital with convulsive movements and altered consciousness. She was born at 33 weeks of gestation with 3300 g birth weight as a first pregnancy of non-consanguineous parents and that she had fever, abdominal swelling and vomiting for six days. On physical examination, she was lethargic and had abdominal distention, mild hepato-splenomegaly, and ascites. Fundoscopic examination was normal. Laboratory analysis showed; Hb 11 g/dL, leukocytes 11300/mm3, platelets 31000/mm3, alanine transferase (ALT) 57 U/L, aspartate transferase (AST) 148 U/L, alkaline phosphatase (ALP) 450 U/L, total bilirubin 3 mg/dL, conjugated bilirubin 1.5 mg/dL, ferritin 20000 ng/mL, lactate dehydrogenase (LDH) 2243 mg/dL, total cholesterol 83 mg/dL (N ≤60), triglyceride 158 mg/dL (N = 32-99), d-dimer 368 mg/dL, total protein 4.7 g/dL, albumin 3.1 g/dL, blood ammoniac 109 µg/dL (N = 19-94) and blood lactate 5.03 mmol/L (N = 0.5-2.2). Urinanalysis, blood glucose, serum electrolytes, amino acids, and amylase were normal. Prothrombin time (PT) and partial thromboplastin time (PTT) were elevated. Fibrinogen was lower than measurable levels. Direct coombs test was negative. Analysis of cerebrospinal fluid (CSF) revealed normal findings. Blood, urine, feces, and CSF were sterile. Serology for cytomegalovirus, Epstein-Barr virus, rubella, toxoplasmosis, parvo-virus, Human 1mmunodeficiency Virus and syphilis were all negative. Brain computed tomography (CT) was normal. Abdominal CT and ultra-sonography revealed hepatomegaly (90 mm), splenomegaly (90 mm), and ascites; there was no solid mass. Atypical mononuclear cells were found in peripheral blood, and hemophagocytosis in the bone marrow smear (Fig. 1).
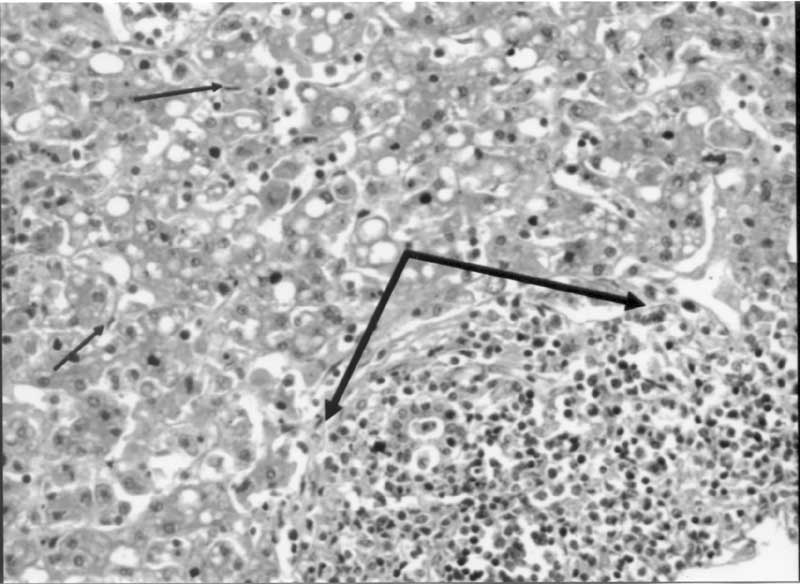
Intravenous immunoglobuline was administered because of progressively declining hemoglobin and platelet counts. Fresh frozen plasma, vitamin K. factor VII, AT-III, platelet and erythrocyte suspensions were administered for coagulopathy. Spironolactone was given for ascites. However, abdominal distention, ascites, and hepatosplenomegaly (below the midclavicular costal margin 7 and 10 cm, respectively) progressed. Later, 100 mL yellow-colored ascitic fluid was obtained by paracentesis. The characteristics of ascitic fluid were; LDH 974 U/L, glucose 61 mg/dL, protein 0.3 g/dL, amylase 9 U/L, cholesterol 62 mg/dL, triglyceride 133 mg/dL, 60 × 10 erythrocytes/mm3, 4 × 10 leukocytes/mm3, cyto-logic examination was normal. Gram stain was negative, and no microorganism was isolated. Paracentesis was repeated three times in two days (100, 200, and 500 mL each time) due to respiratory distress. At the sixth day of referral, a bleeding was seen from her mouth and noise, requiring mechanical ventilation. ALT and AST levels were 437 U/L, and 3925 U/L, respectively. At the same time she became unconscious and later died. Histopathologic findings of necropsy were as follows: Hepatocytes were damaged severely, and Kupffer cells were increased. Throughout the portal areas, there were CD8+ stained mononuclear cell infiltrations destroying the hepatocytes (Fig. 2). These infiltrative areas were stained as S-100- and CDla-. Iron stain was positive in the liver. Spleen and lymph node were also infiltrated by CD8+, CD57+, and CD68+ cells.
Discussion HLHs are characterized by accumulation of antigen-processing cells (i.e., macrophages) in many organs. With the characteristic morphology of normal macrophages by light microscopy, these phagocytic cells lack the two markers (Birbeck granules and CDla positivity) characteristic of the cells found in Langerhans cell histiocytosis(3,6). Clinical and laboratory criteria for the diagnosis of HLH are; (i) fever and splenomegaly, (ii) cyto-penia in 2 of 3 lineages, (iii) hypertriglyceridemia or hypofibrinogenemia or both, (iv) hemophagocytosis in the bone marrow, spleen, or lymph nodes, and (v) absence of malignancy(1-3). Hyperlipidemia is an important feature of this condition and may help in the diagnosis(2,3). She had elevated levels of cholesterol and triglycerides. Additionally, the ascitic fluid contained increased levels of lipids. In patients with HLH, the ferritin levels may increase, so the disease may be misdiagnosed as neonatal iron storage disease(2). Neurologic involvement may also be present, manifesting as irritability, convulsions, meningitis, and altered consciousness(2,3). The convulsive movements, lethargy, and altered consciousness of our patient suggest neurologic infiltration. In most cases, hepatic injury occurs with elevated liver enzymes and cholestasis. Up to our knowledge, ascites is reported only in two cases with HLH (one with FEL and other with IAHS) in the literature(4,5). The patient suffering from FEL was an eight month-old boy(5). We could not find any case as young as our patient in the literature. The cause of ascites may be due to the infiltrations of lymphatic system with lymphohistiocytes, more than liver damage. Untreated familial disease is fatal in all cases. Also acquired IAHS has a high fatality rate of 50% in children. If a treatable organism is found appropriate therapy should be given. The immediate aim of treatment is to suppress hypercytokinemia that is responsible for the life-threatening symptoms. Standard treatment for HLH is a combination of corticosteroids, cyclosporin A and etoposide. All patients with known familial disease suspected genetic disease due to age below 1 year and patients with life-threatening symptoms such as coagulopathy, profound cytopenia or neurological disease should receive therapy. In familial cases this has to be followed by stem cell transplantation (SCT) as the only curative disease(7). Contributors: MA, RA, AY responsible for patient follow up and treatment; BA responsible for histopathological examination. Competing interests: None. Funding: This study was supported by Akdeniz University Scientific Research Project Unit.
| ||||
|
References | ||||
|
|
![]()