|
|
Case Reports Indian Pediatrics 2006; 43:434-437 |
|||||
|
Glomerulocystic Disease |
|||||
|
Correspondence to: Dr. M. Vijayakumar, Consultant
Pediatric Nephrologist, Kanchi Kamakoti CHILDS Trust Hospital, 12-A,
Nageswara Road, Numgambakkam, Chennai 600 034, Tamilnadu, India.
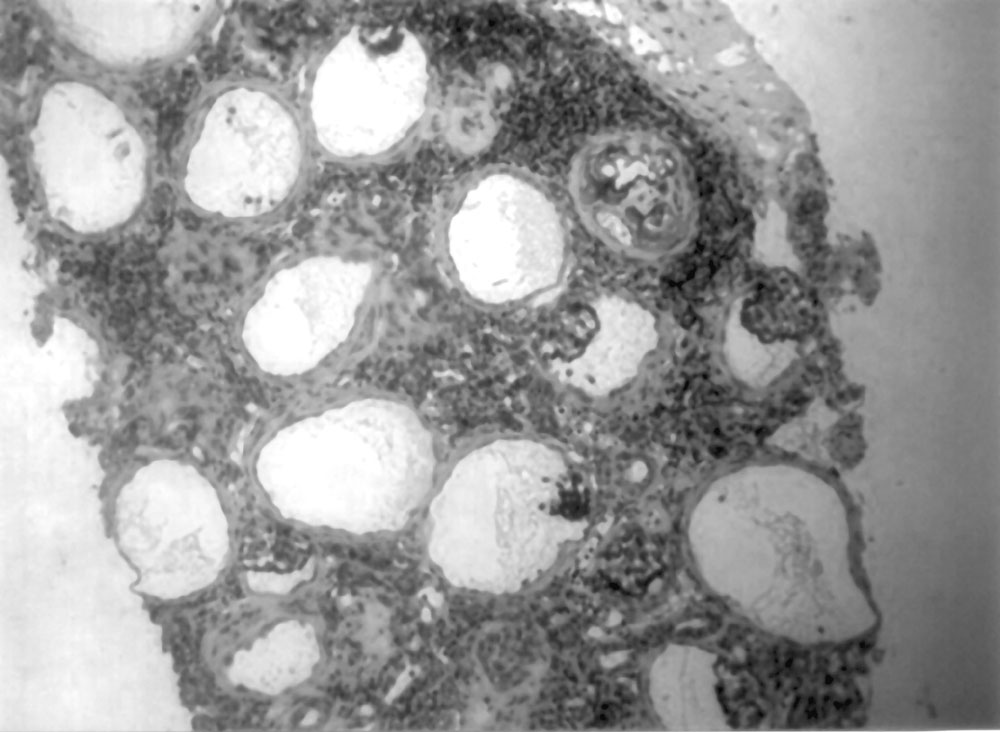
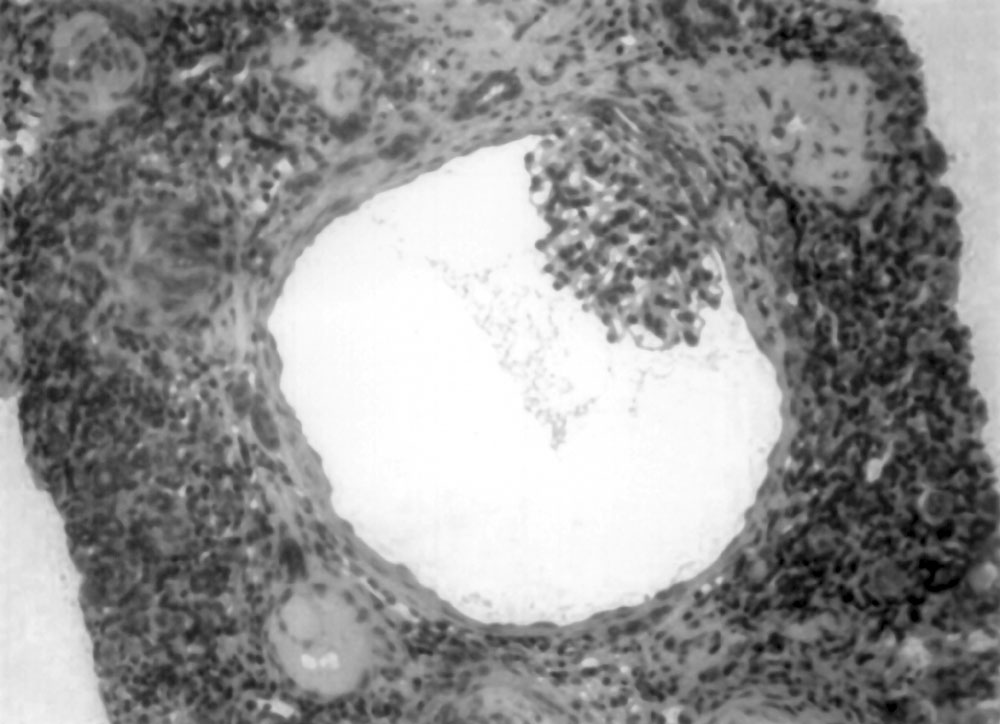
Various renal cystic diseases including autosomal dominant polycystic kidney disease (ADPKD) present with variable renal dysfunction from neonatal to adolescence. Glomerulocystic kidney disease (GCKD) is a different entity characterized by glomerular cysts and associated renal dysfunction. GCKD is a rare condition with both sporadic and familial occurrence, characterized by cystic dilatation of Bowman’s space and occasionally the initial proximal convoluted tubule. It can be classified further based on presentation, size of the kidneys and associated multisystem disorders. We report a young child with GCKD. Case Report A 9-month-old girl, born to non-consanguineous parents, with normal ante-natal ultrasound and no history of renal disease in the family was noticed to have occasional vomiting since birth. Gradually her vomiting increased associated with diarrhea at 4 months of life. On evaluation, she was found to have serum creatinine level of 3.3 mg/dL and potassium of 4.2 mEq/L. Ultrasonogram showed left pelvic kidney and normally placed right kidney, which were of normal size with increased echogenecity and loss of cortico-medullary differentiation. She was managed with IV fluids and peritoneal dialysis for 4 days at the local center. She improved symptomatically but had persistently elevated serum creatinine of 1.4-1.8 mg/dL. At 9 months, she weighed 5.25 kg, was anemic with no dysmorphic features. She was well hydrated but had acidotic breathing; blood pressure level was 80/60 mm Hg. Investigations showed serum creatinine of 2.0 mg/dL, sodium 143 mEq/L, potassium 4.6 mEq/L, chloride 115 mEq/L, bicarbonate 11 mEq/L, total protein 7.9 g/dL, albumin 3.9 g/dL, cholesterol 334 mg/dL, calcium 9.0 mg/dL, phosphorus 2.7 mg/dL, alkaline phosphatase 1583 IU/L, uric acid 13 mg/dL, hemoglobin 6.1 g/dL and PCV 17%. Urinalysis showed specific gravity of 1.010, pH 5.0, albumin 3+ and occasional RBCs. The random urine protein was 397 mg/dL and creatinine 10 mg/dL; protein to creatinine ratio 39.7. Ultrasound examination confirmed the previous findings. Micturating crystourethrogram showed no evidence of VUR. The patient was managed with oral sodium bicarbonate (5 mL three times a day) and oral allopurinol (25 mg twice a day). She received packed cell transfusion, followed by subcutaneous injections of erythropoietin (250 IU weekly), oral iron (5 mg/kg/day) and folic acid (2.5 mg/day). An ultrasound guided renal biopsy was done on the right kidney under general anesthesia using biopty gun. Renal histopathology showed 20 glomeruli with marked dilatation of the Bowman’s capsule with collapsed capillary tuft projecting into the lumen covered by a prominent layer of visceral epithelial cell (Figs. 1 & 2). There were also ischemically obsolescent small glomeruli and blood vessels showing fibro-intimal hyperplasia and luminal narrowing. Interstitial infiltrations of lymphocytes were noted. Some dilated and atrophic tubules were present focally and focal calcification of suspected glomerular structures were seen. Immunofluorescence examination showed insignificant deposits of IgM and C3c in the glomeruli. The histopathology was suggestive of GCKD. The patient was discharged on treatment with oral sodium bicarbonate solution, allopurinol, iron and folate supplements and weekly subcutaneous injections of erythropoietin. On follow-up at 11 months of age the patient weighed 7.4 kg with near normal motor development and serum creatinine level of 1.4 mg/dL.
Discussion GCKD represents a heterogeneous collection of heritable and non-heritable clinical entities and is diagnosed by typical histopathology(1). There are 3 main groups of GCKD: (a) non-syndromal heritable and sporadic form of GCKD in children and adults, (b) GCKD as a major component of heritable malformation syndromes, and (c) glomerular cyst as a minor component of abnormal or dysplastic kidney disease, some of which are syndromal. GCKD with isolated renal involvement may be inherited in an autosomal dominant manner or can occur as a familial hypoplastic disease or as a sporadic occurrence. Reports of kindreds with GCKD suggest autosomal dominant inheritance in some case. Studies have shown that the distinctive GCKD pheno-type in some families results from a dominantly acting mutation that disrupts a genetic locus distinct from the ADPKD loci(2). Hypoplastic GCKD is a distinct clinical entity, which is inherited dominantly and is reported only in few families(3,4). These kidneys apart from being glomerulocystic are small with abnormal pyelocalyceal anatomy. Recently, mutation in the hepatocyte nuclear factor-1beta gene has been identified in hypoplastic GCKD variant(5). GCKD may be associated with the orofacial digital syndrome type 1, brachymeromelia renal syndrome, trisomy-13, Majewiski type short rib polydactyly syndrome and nephronopthisis. Although tubular cyst may be seen in tuberous sclerosis, commonly it may be associated with GCKD. Syndromes associated with glomerular cyst as a component of renal dysplasia includes Meckel syndrome, glutaric aciduria type 2 and renal hepatic pancreatic dysplasia(6). It is difficult to distinguish GCKD from other cystic diseases. In GCKD, the kidneys are enlarged due to cysts(6), which are dilated Bowman’s spaces comprising a sphere lined with cuboidal or columnar cells and containing abortive or primitive glomeruli, separated by normal parenchyma(1). The cysts are normally in the cortex, sparing the medulla. The tubules are usually spared which distinguishes it from other cystic diseases. Rarely, GCKD may be associated with tubular cyst and dysplasia(6). GCKD may present in infancy with abdominal masses and renal failure. Later they might present with hypertension, flank pain and hematuria. The earlier the diagnosis is made, more severe the course of the disease. Patients with familial hypoplastic GCKD variant have small kidneys with abnormal collecting system and abnormal or absent papillae and they present with chronic renal failure in early life, but tend to have stable course later on. Ultrasonography demonstrates bilateral renal enlargement without distortion of renal contour with increased cortical and medullary echogenecity with loss of cortico-medullary differentiation and evidence of cortical cyst. The main differentiating feature to distinguish GCKD and ARPKD is abnormal medullary pyramids in the later. In future, CT scan and MRI may be valuable in distinguishing these two diseases. The clinical course and prognosis are quiet variable and dependant on the presence of associated disorders. Acknowledgment We thank the Department of Histopathology, Apollo Hospitals, Chennai for their help in histopathology. Contributors: MVK, NP and BRN managed the child. MVK and NP wrote the manuscript and BRN reviewed it. MVK shall act as the guarantor. Funding: None. Competing interest: None stated.
| |||||
|
References | |||||
|
|
![]()