A 15-month-old child was brought for evaluation of ambiguous genitalia
and failure to thrive. He was irritable, underweight and short for age.
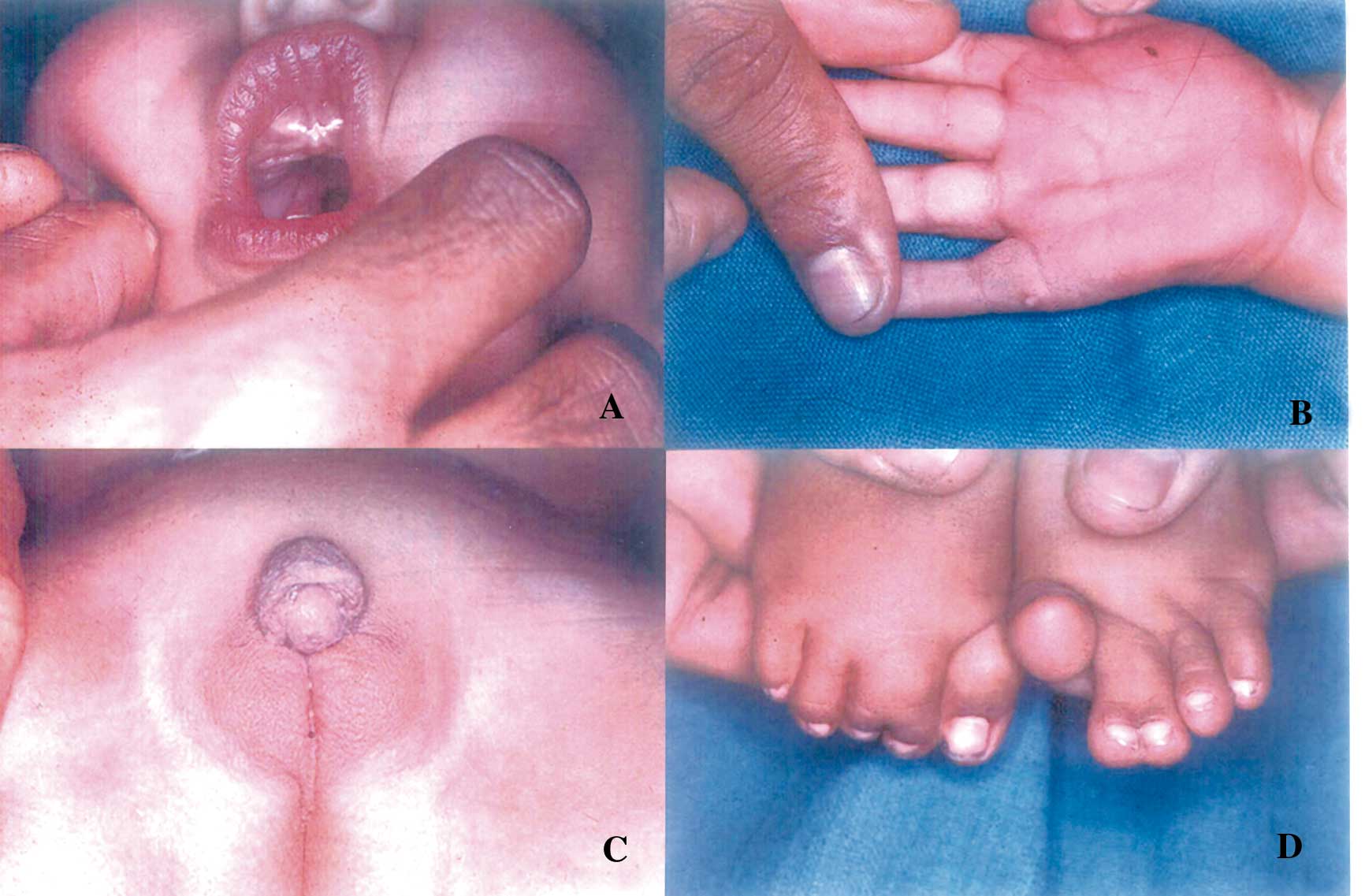
He had hypopigmented hair, hypertonia, scissoring of lower limbs,
asymmetrically short flexed fingers, micro-cephaly, narrow frontal area,
slanting auricles, broad nasal tip, anteverted nostrils, microg-nathia,
cleft palate, thickened dental ridge, simian crease, polydactyly,
metatarsus abductus, syndactyly of second and third toes, and ambiguous
genitalia (micropenis, hypo-spadias, cryptorchidism and a rudimentary
vagina with a karyotype of 46XY) (Fig. 1). The child had
pyloroplasty for congenital hypertrophic pyloric stenosis at 28 days of
age. An ultrasound examination of abdomen revealed left sided
hydronephrosis, duplex collecting system, ureterocele and cystic
dysplasia of upper calyceal system. His lipid profile revealed low serum
cholesterol levels and absent LDL cholesterol. A diagnosis of Smith-Lemli-Opitz
syndrome was made based on the clinical and biochemical profile.
 |
|
Fig. 1. A. Showing cleft palate and thickened
alveolar ridges, B. showing
polydactyly and simian crease. C. showing ambiguous genitalia and
D. showing
syndactyly of second and third toes. |
Smith-Lemli-Opitz syndrome, first des-cribed in 1964,
is a rare autosomal recessive disorder of cholesterol synthesis wherein
the conversion of 7-dehydro-cholesterol (7-DHC) into cholesterol is
disrupted leading to excessive accumulation of 7-dehydro-cholesterol,
8-dehydro-cholesterol (8-DHC, an isomeric form of 7-DHC), and deficiency
of cholesterol. The accumulated 7-DHC and 8-DHC also cause decreased
HMG-CoA reductase activity. Hence cholesterol replace-ment forms the
cornerstone in medical management of this condition. The coexisting
surgical conditions may need appropriate evaluation and intervention.
Julius Xavier Scott,
Praburam P.M.,
Department of Child Health,
Christian Medical College & Hospital,
Vellore,
India.
E-mail: [email protected]
