|
Case Reports Indian Pediatrics 2000;37:542-545 |
||||||||||
|
Aicardi’s
Syndrome in a Male Child: An Unusual Presentation
|
||||||||||
|
K.C.
Aggarwal
Archana Aggarwal* M.S. Prasad R.N. Salhan Amit Upadhaya
In 1965, Aicardi and colleagues reported a new syndrome characterized by agenesis of corpus callosum (ACC) with cortical heterotopia, infantile spasm, chorioretinopathy, mental retardation with or without associated vertebral anomalies. Amongst these, ACC, infantile spasms, mental retardation and chorioretinal lacunae are the constant findings(1). It is a rare neuro-ophthalmic disorder with progressive mental deterioration. All the patients described till date have been females except two male subjects(2,3). We describe a male baby who had all the essential features suggestive of Aicardi’s Syndrome along with ventricular septal defect and lissencephaly.
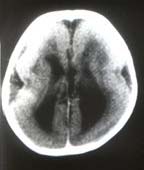
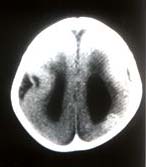
A 9-month-old male child, first in birth order was a product of full term normal vaginal delivery with normal perinatal period. No history of consanguinity was noted. The child presented to us with delayed milestones, recurrent recalcitrant infantile spasms, inability to recognize his mother and history suggestive of recurrent chest infections (poor feeding and fever with fast respiration). No family member was affected with a similar illness and there was no history suggestive of intrauterine infection in the antenatal period. On examination, there was microcephaly with head circumference of 37 cm (less than –3 SD). There was no evidence of cranio-synostosis or neurocutaneous malformation. The cardiovascular examination revealed Grade III ejection systolic murmur best heard at left parasternal border, flow middiastolic rumble and S3 at the apex. P2 was loud with normal split. Chest examination revealed tachypnea with subcostal retractions and basal crepita-tions. Central nervous system examination showed a hypotonic child with brisk deep tendon jerks with extensor plantars. Head control was partial with some head lag. The child responded to sound. Vision was doubtful with left side micro-opthalmia. The fundus examination, done under general anesthesia revealed bilateral chorioretinal lacunae, which were more prominent on the right side. No evidence of chorioretinitis was noted. There was no hepatosplenomegaly. Investigations revealed cardiomegaly with increased pulmonary vascularity on X-ray chest. Electrocardiogram revealed left ventricular preponderance with normal QRS axis and tachycardia. Color Doppler Echocardiography confirmed moderate to large subaortic VSD with pulmonary blood flow more than 2:1. EEG showed diffuse background slowing with asynchronous burst-suppression pattern. Brain stem auditory evoked responses were normal. Visually evoked potential showed abnormal record. Investigation for TORCH group of infections was negative. X-ray skull did not show any calcification. CT scan head showed agenesis of corpus callosum with bat-wing anomaly with lissencephaly, i.e., smooth surface of cerebral cortex with absence of gyri and sulci (Figs. 1&2). MRI confirmed the CT head findings and also showed heterotopia with migration defect. Skeletal survey of baby did not reveal any abnormality except mild thoracic scoliosis.
In the present case all the essential features suggestive of Aicardi’s Syndrome, i.e., infantile spasms with agenesis of corpus callosum, chorioretinal lacunae and micro-ophthalmia were present. No other skeletal anomaly was detected except thoracic scoliosis. Since the first case description, only two males with this disorder have been reported earlier(2,3). Karyotyping is not recommended routinely in this condition. However, because of phenotypic male sex, it was performed in this patient. The karyotype was 46 xy but mosaicism can not be excluded. Isolated
agenesis of corpus callosum (ACC) can be a chance finding on CT Scan
head. Majority of them remain asymptomatic throughout childhood. Many
syndromes have been documented with ACC but the most widely described
one is Aicardi’s syndrome in which ACC is associated with neuronal
migration defect in the cerebral cortex, especially in periventricular
area. In addition to this, polymicrogyria and neuronal heterotopia
may also be associated. Lissen-cephaly, i.e., smooth surfacing
of cerebral cortex with absence of sulci and gyri has not been described
in Aicardi’s syndrome earlier and is another unusual feature in this
case. A close differential diagnosis is Walker Waburg syndrome (WWS)
which is comprised by lissencephaly (type 11), microophthalmia, retinal
dysplasia and hypotonia. The described case does not fit into WWS
as there is no absence of cerebellar vermis, there is extreme type
of lissencephaly (type 1) and ACC is present. Miller Dicker syndrome,
which is also associated with lissencephaly and heart defect can be
excluded by the presence of retinal changes and ACC(4).
|
||||||||||
![]()