|
|
Case Reports Indian Pediatrics 2001; 38: 292-294 |
||||||||||
|
Autosomal
Recessive Polycystic Kidney Disease with Congenital Hepatic |
||||||||||
|
Autosomal recessive polycystic kidney disease (ARPKD) is genetically distinct from adult polycystic kidney disease(1). Depending on time of presentation and presence of associated hepatic lesions, perinatal, neonatal, infantile and juvenile sub-categories have been described. The association of autosomal recessive polycystic kidney disease with congenital hepatic fibrosis is well known(2). But a rare occurrence is that of encephalo-cele, bilateral congenital talipes equinovarus with autosomal recessive polycystic kidney disease. We report an infant with these associations.
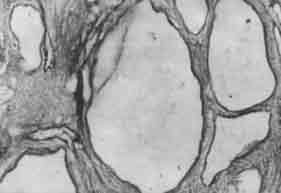
A preterm female baby, was delivered at 36 weeks gestation weighing 1.9 kg with an APGAR score of 3/10 at 1 min and 5 min. The baby died after 20 min of resuscitation. The parents were cosanguineous. An antenatal diagnosis of multicystic kidney disease and encephalocele had been made on ultrasound. Clinical examination of the newborn revealed bilateral renal masses, congenital talipes equinovarus (CTEV) and occipital encephalo-cele. Autopsy findings revealed enlarged kidneys weighting 70g each. The outer surface of the kidneys was bosselated and cut section showed multiple cysts varying in size from 0.5 to 1.5 cm involving the cortex and medulla. More than 90% of the parenchyma was filled with cysts. The pelvicalyceal system was poorly demarcated. Histopatho-logical examination revealed infantile type of polycystic kidney (Fig. 1). Liver was enlarged and weighed 140 g. The outer surface of the liver was nodular. Cut-sections showed grey-brown areas of fibrosis confirmed by histopathological examination (Fig. 2). The presence of congenital talipes equinovarus (CTEV) and encephalocele were confirmed. Karyotyping revealed normal 46 XX chromosomes.
Polycystic diseases are a diverse group of heritable disorders often associated with renal and hepatic cystic lesions. The cystic diseases of the kidney are congenital or acquired. Two basic abnormalities necessary for cyst forma-tion are increased epithelial cell proliferation and altered fluid transport. Congenital polycystic kidney are of two types, i.e., autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease. ARPKD may often end with either stillbirth or death during infancy. Autosomal dominant polycystic kidney disease is relatively more common and has good prognosis. Congenital hepatic fibrosis (CHF) is an autosomal recessive disease presenting princi-pally in childhood and often associated with renal manifestations(3) like cystic disease of kidney. Portal hypertension and increased risk of ascending cholangitis are commonly associated. The association of ARPKD with congenital hepatic fibrosis (CHF) is well known. When the severity of hepatic mani-festations exceeds renal involvement the disorder is called CHF(4,5). In the present case there were bilateral flank masses at birth suggestive of ARPKD. The association of bilateral CTEV and encephalocele make this case unusual. Meckel Gruber syndrome is an autosomal recessive condition, characterized by occipital encephalocele, polycystic kidneys and poly-dactyly. A wide phenotypic expression of Meckel Gruber syndrome is reported. How-ever, CHF is not described in Meckel Gruber syndrome. CTEV may be associated with renal diseases due to oligohydramnios. The presence of CTEV in our case could be because of severe renal pathology. The association of ARPKD, CHF, posterior encephalocele and bilateral CTEV is not described earlier and thus make this case unique.
Contributors:
SK and RVB conducted autopsy and prepared the manuscript. BVB helped to
prepare the manuscript.
|
![]()