|
|
|
Indian Pediatr 2014;51: 203-210 |
 |
Management of Neonatal Cholestasis: Consensus
Statement of the Pediatric Gastroenterology Chapter of Indian
Academy of Pediatrics
|
|
Vidyut Bhatia, *Ashish Bavdekar,
$John
Matthai, #Yogesh Waikar and
Anupam Sibal
From Apollo Center for Advanced Pediatrics,
Indraprastha Apollo Hospital, New Delhi; *Department of Pediatrics, King
Edwards Memorial Hospital, Pune; $Department of Pediatrics, PSG
Institute of Medical Sciences, Coimbatore and #Pediatric
Gastroenterology Clinic, Care hospital, Nagpur, India.
Correspondence to: Dr Vidyut Bhatia, Apollo Center for
Advanced Pediatrics, Indraprastha Apollo Hospital,
New Delhi 110 076, India. Email: [email protected]
|
Justification:
Neonatal cholestasis is an important cause of chronic liver disease
in young children. Late referral and lack of precise etiological
diagnosis are reasons for poor outcome in substantial number of
cases in India. There is a need to create better awareness among the
pediatricians, obstetricians and primary care physicians on early
recognition, prompt evaluation and referral to regional centers.
Process: Eminent
national faculty members were invited to participate in the process
of forming a consensus statement. Selected members were requested to
prepare guidelines on specific issues, which were reviewed by two
other members. These guidelines were then incorporated into a draft
statement, which was circulated to all members. A round table
conference was organized; presentations, ensuing discussions, and
opinions expressed by the participants were incorporated into the
final draft.
Objectives: To review
available published data on the subject from India and the West, to
discuss current diagnostic and management practices in major centers
in India, and to identify various problems in effective diagnosis
and ways to improve the overall outcome. Current problems faced in
different areas were discussed and possible remedial measures were
identified. The ultimate aim would be to achieve results comparable
to the West.
Recommendations: Early
recognition, prompt evaluation and algorithm-based management will
improve outcome in neonatal cholestasis. Inclusion of stool/urine
color charts in well baby cards and sensitizing pediatricians about
differentiating conjugated from the more common unconjugated
hyperbilirubinemia are possible effective steps. Considering the
need for specific expertise and the poor outcome in sub- optimally
managed cases, referral to regional centers is warranted.
Keywords: Cholestatic jaundice, Neonate,
Practice guidelines.
|
|
Neonatal cholestasis (NC) is being increasingly
recognized as an important cause of chronic liver disease in infants and
young children. The etiology and management of cholestasis has changed
significantly since the consensus statement last published in 2000 [1].
Three objectives were identified at the previous meeting. First, age at
which infants are referred to tertiary care centers is very late for
effective evaluation and management and steps need to be taken to
address this problem. Second, tertiary centers with pediatric
gastroenterology units should follow a uniform protocol of evaluation,
and third, the investigation facilities at some centers need to be
strengthened, so that a more precise final diagnosis, particularly
metabolic disorders, can be arrived at.
A number of steps including a public campaign on
‘Yellow alert’ and educational programs at pediatric meetings were since
held to address the key issues [2,3]. The algorithm put in place in 2001
was followed at almost all major centers, until newer advances were
reported. Laboratories in many private and other medical centers are now
able to do newer tests for precise diagnosis. This meeting was held to
discuss the impact of those programs and to make appropriate changes in
the management protocol in the light of recent advances in the subject.
Definition
Neonatal cholestasis is defined as conjugated
hyperbilirubinemia occurring in the newborn as a consequence of
diminished bile flow. Conjugated hyperbilirubinemia in a neonate is
defined as a serum direct/conjugated bilirubin concentration greater
than 1.0 mg/dL if the total serum bilirubin (TSB) is <5.0 mg/dL or
greater than 20 percent of TSB if the TSB is >5.0 mg/dL [4]. It is
important to note that the diazo method of estimating bilirubin, that is
still practiced in many Indian centers, tends to overestimate the direct
fraction at lower bilirubin levels. The group however felt that the
above mentioned definition has to be retained since it is an
internationally accepted one.
Conjugated hyperbilirubinemia at any age in a newborn
is pathological and requires evaluation. Any newborn with jaundice and
dark yellow urine staining the diaper with or without pale stools should
be strongly suspected to have NC. Such babies must be referred to an
appropriate center for further investigations and treatment at the
earliest [1]. Sick newborns and younger infants with deranged liver
function tests, particularly uncorrected coagulopathy, require urgent
referral to rule out infective or metabolic causes of NC.
Etiology
NC affects 1 in 2500 infants in the West [4,5]. In
India it constitutes 19% to 33% of all chronic liver diseases in
children reporting to tertiary care hospitals [1,6-8]. Table I
summarizes the etiologic profile of NC in India. Hepatocellular causes
constitute 45% to 69% while obstructive causes account for 19% to 55% of
all cases [1,6-12].
TABLE I Etiologic Profile of Neonatal Cholestasis in India
|
Etiologic Factor |
N-1008* (%) |
N-420#(%) |
|
Obstructive causes |
|
|
|
Biliary atresia |
34 |
30 |
|
Choledochal cysts |
4 |
5 |
|
Hepatocellular causes |
|
|
|
Infections |
17 |
18 |
|
Metabolic causes |
4 |
12 |
|
Miscellaneous |
2 |
3 |
|
Unknown etiology |
30 |
31 |
|
Ductal paucity |
3 |
1 |
|
Undifferentiated |
6 |
1.2 |
*Based on cumulative data from eight
tertiary care centers [1];
#Data from the largest single series (7). |
While 20 to 30% of cases of NC were idiopathic in
earlier studies, [1,6,7,9], recent reports documented this proportion to
be lower [2,8,13-18]. The group felt that this emphasizes the need for
more exhaustive work up than was recommended earlier. A recent
publication with exhaustive workup has shown that Pi-Z and Pi-S alleles
responsible for alpha-1 antitrypsin deficiency may be rare in our
population [7].
Clinical Presentation
Jaundice in newborns is most commonly physiological
or due to ABO/Rh hemolytic incompatibility. However, if jaundice is
associated with dark urine and/or pale stools, it is suggestive of
cholestasis. The sensitivity, specificity, and positive predictive value
of pale stools for the detection of biliary atresia (BA) before 60 days
as determined by a color-coded stool chart was noted to be 89.7%, 99.9%
and 28.6%, respectively [19]. Although cholestasis is known to occur in
babies with early onset sepsis [14]. Yet simultaneous screening should
be done for metabolic causes. Galactosemia, a treatable cause, may be
present even in babies with culture proven sepsis. Babies with
tyrosinemia, herpes infection and congenital hemochromatosis may present
in a sick state early in life, and their recognition and treatment needs
to be incorporated [20]. Non-normalization of liver function tests
(LFTs) even after treatment with appropriate antibiotics is a pointer
towards an underlying metabolic cause for sepsis [14].
Yachha, et al. showed that the age of onset of
jaundice in BA was 3-12 days and that of hepatocellular causes was 16-24
days [2]. However, the mean age of presentation to a tertiary care
center was 2.8–3.9 months compared to the desired age of evaluation,
that is 4-6 weeks [2,3,15]. Babies with BA appear well and have normal
growth and development in spite of their jaundice, and this leads to
parents and physicians underestimating the seriousness of the problem
[3]. Many health care professionals also have a misconception that all
well babies with icterus have physiological jaundice (which is
unconjugated and associated with normal urine color). The group felt
that it was a matter of concern that in India the average age of
presentation to tertiary care centers has shown little change in the
last decade. The group felt that there is a need for creating more
awareness among pediatricians and obstetricians on NC and this can be
achieved through continuing medical education (CME) programs with the
speakers giving the same message by highlighting the recommendations of
this consensus statement. Enlarging the scope of the ‘yellow alert’ to
include the whole country through visual and print media would also be
beneficial. The Group recommended urine and stool color assessment
(minimum 3 stool samples) by the mother and physician in a stool color
card incorporated in all well baby cards (Indian Academy of Pediatrics
and the Government of India cards). The group also felt that the
Taiwanese experience with stool color cards (subsequently replicated in
other countries) should be possible in India as well [21].
Persistent cholestasis from any cause leads to liver
damage and cirrhosis. Therefore, determining the specific etiology
(medical or surgical) at the earliest is critical. Also, the outcome of
Kasai portoenterostomy (PE) is directly related to the age of surgery
and the expertise of the treating unit. PE when performed earlier than
60 days of age established adequate bile flow in 64.7% of patients
compared with 31.8% when performed later [22,23]. Improved outcome is
also noted in centers with a higher case load [24]. In the United
Kingdom, a center performing >5 Kasai PE per year, reported
significantly better outcome [25]. Hence, babies suspected to have BA
require early referral to an appropriate center with expertise in
performing PE.
Investigations
Most centers have been following the protocol that
was outlined in the earlier consensus statement. However in the light of
recent publications and better laboratory support, modifications are
required in the evaluation and management protocol. The group felt that
laboratory facilities and histopathology support has improved
tremendously in the last decade and hence better work up is now possible
in many centers. The initial evaluation of an infant with NC includes a
complete liver function test (LFT), thyroid function test, and a sepsis
screen followed by specific radiological and histopathological tests
(Fig. 1). The principal diagnostic concerns are to
differentiate hepatocellular diseases from anatomical disorders, and
diseases that are managed medically from those requiring surgical
intervention. The most important initial investigation is to establish
cholestasis by measuring serum bilirubin (total and differential)
levels. Severity of liver dysfunction can be measured by estimating the
prothrombin time or international normalized ratio (INR) and serum
albumin. No single laboratory or imaging test exists which
differentiates biliary obstruction from other causes of NC reliably in
all cases. The serum transaminases are sensitive indicators of
hepatocellular injury, but lack specificity and prognostic value. High
alkaline phosphatase levels can be seen in biliary obstruction, but has
very low specificity. Gamma-glutamyl transpeptidase (GGTP) is a marker
of biliary obstruction and is elevated in most cholestatic disorders;
paradoxically low or normal levels are found in patients with
progressive familial intrahepatic cholestasis (PFIC) and disorders of
bile acid synthesis [26].
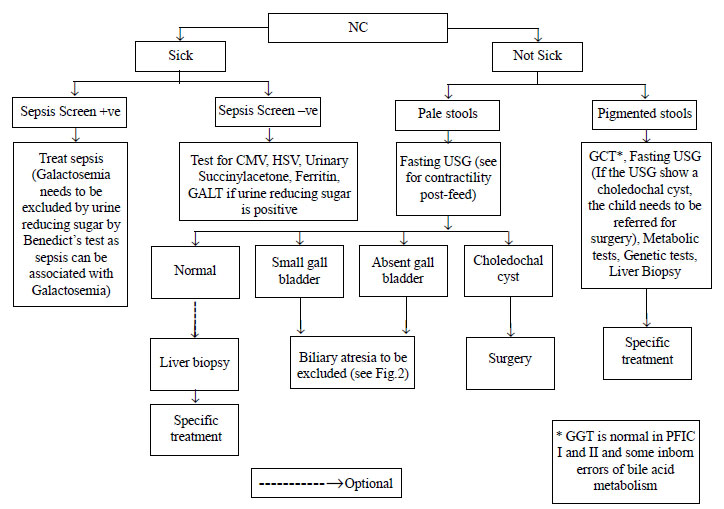 |
|
Fig. 1 Diagnostic algorithm for
management of neonatal cholestasis (Part 1).
|
Abdominal ultrasonography may provide findings
suggestive of BA and can also be used to confirm the existence of other
surgically treatable conditions like choledochal cyst, inspissated bile
plug syndrome and choledocholithiasis. Abdominal ultrasonography
findings described in BA include the triangular cord sign [27], abnormal
gallbladder morphology (not visualized or length <1.9 cm or lack of
smooth/complete echogenic mucosal lining with an indistinct wall or
irregular/lobular contour) [19], no contraction of the gallbladder after
oral feeding and non-visualized common bile duct (CBD). A distended gall
bladder, however, does not rule out a proximal BA with a distal patent
bile duct and mucus filled gallbladder. It is recommended that
ultrasound should be done after 4 hours of fasting.
Hepatobiliary-imino-di-acetic acid (HIDA) scan has
limited role in evaluation of NC especially if the baby has clearly
documented pale or pigmented stools. The time required (5-7 days) for
priming before the scan, especially in patients who are referred late,
is a limitation. The group felt that good-quality HIDA scan may not be
available everywhere in our country, and therefore, delaying the liver
biopsy for it is not justified. Performing a HIDA scan is optional and
one may go for a liver biopsy straightaway (Fig. 2). HIDA
is, however, useful in the diagnosis of the uncommon causes like
spontaneous perforation of the bile duct [28]. Intra-operative
cholangiogram (IOC) remains the gold standard for diagnosis of BA.
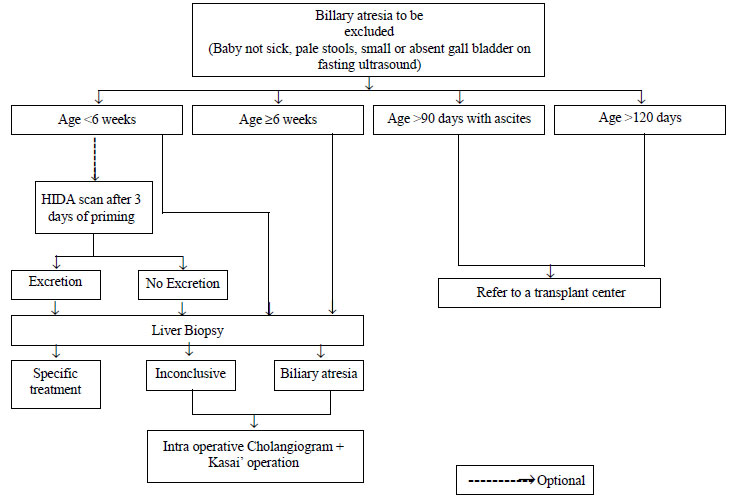 |
|
Fig. 2 Diagnostic algorithm to help in
the management of neonatal cholestasis (Part 2).
|
Liver biopsy is an essential investigation in the
evaluation of NC. Early recognition of BA by liver biopsy can avoid
unnecessary laparotomy (5). The characteristic histopathology features
of BA are bile duct proliferation, bile plugs in ducts, fibrosis and
lymphocytic infiltrates in the portal tracts. The reported sensitivity,
specificity and accuracy of liver biopsy in the diagnosis of BA are
89-99%, 82-98% and 60-95%, respectively [16]. Percutaneous liver
biopsy in early infancy under local anesthesia and sedation is a safe
procedure, if performed by competent physicians [29]. The group opined
that the biopsy must be interpreted by an experienced pathologist in
conjunction with the clinical profile and results of the other
investigations. In cases of doubt, a second opinion should be sought.
In galactosemia, urine is positive for non-glucose
reducing substances while the infant is on lactose feeds. E. coli
sepsis in the presence of liver cell dysfunction is very characteristic
of galactosemia. Assay of Galactose-1 phosphate uridyl transferase
(GALT) enzyme is used for confirmation. Mutational analysis of the GALT
gene from Indian subjects has revealed heterogeneity in the structure of
the gene and the presence of novel mutations [30,31]. Hereditary
fructose intolerance (HFI) is not an uncommon cause of neonatal
cholestasis in our country as there are many states where sugar water
continues to be given to newborns until lactation is fully established.
It should be considered in clinical settings where sucrose or fruit
juices have been given to babies. Assay of aldolase B enzyme in liver
biopsy sample confirms HFI. Fructose challenge test can make the child
very ill and is now obsolete. Plasma tyrosine levels are unreliable in
the diagnosis of tyrosinemia. Measurement of urinary succinylacetone and
succinyl acetoacetate or assay of the FAH gene is diagnostic [32]. Among
the congenital infections, cytomegalovirus (CMV) is most commonly
implicated. Serum IgM level is unreliable in diagnosis and should not be
used. Assay of pp65 antigen and CMV polymerase chain reaction (PCR) are
more specific and reliable, if the biochemical tests and histology are
consistent with the diagnosis. Markedly raised serum ferritin and
uncorrected coagulopathy are suggestive of hemochromatosis that may be
confirmed by a buccal mucosal biopsy.
A two part diagnostic algorithm to help in the
management of NC is given in Fig.1 and Fig. 2.
Treatment
General Medical Management
Most infants with NC are underweight and will need
nutritional support. The goal is to provide adequate calories to
compensate for steatorrhea and to prevent/treat malnutrition. The
calorie requirement is approximately 125% of the recommended dietary
allowance (RDA) based on ideal body weight [33]. In breastfed infants,
breastfeeding should be encouraged and medium-chain triglyceride (MCT)
oil should be administered in a dose of 1-2 mL/kg/d in 2-4 divided doses
in expressed breast milk [34]. In older infants, a milk-cereal-mix
fortified with MCT is preferred. Adding puffed rice powder and MCT to
milk can make feeds energy-dense. Essential fatty acids should
constitute 2-3% of the energy provided. Vegetable protein at 2-3 g/kg/d
is recommended [34].
Infants with cholestasis require supplementation with
fat-soluble vitamins administered orally as water-soluble preparations.
Suggested daily vitamin and mineral supplementation are given in
Table II. In treatment of vitamin deficiencies, standard
deficiency protocols should be followed. 1,25 dihydroxy Vitamin D3
(0.05-0.2 ug/kg/d) is recommended in the presence of significant bone
changes or patients having severe cholestasis [34]. Vitamin K is
administered at a dose of 5 mg intramuscular, subcutaneously or
intravenously, at diagnosis to correct the coagulopathy. If the INR is
markedly prolonged, intramuscular injections should be avoided. Vitamin
supplementation should be continued till 3 months after resolution of
jaundice [35].
TABLE II Suggested Daily Vitamin and Mineral Requirements in Infants with Cholestasis
|
Route |
Dose* |
|
Vitamin A# |
Oral |
5000-25,000 IU/d |
|
Vitamin D |
Oral |
400-1200 IU/d |
|
Vitamin E |
Oral |
50-400 IU/d or 15-25 IU/kg/d of TPGS form
if available |
|
Vitamin K$ |
Oral |
2.5 twice/week to 5 mg/d |
|
Parenteral |
2-5 mg IM, SC or IV 4 weekly |
|
Water soluble vitamins |
Oral |
1-2 times the RDA |
|
Calcium** |
Oral |
20-100 mg/kg/d |
|
Phosphorus |
Oral |
25-50 mg/kg/d |
|
Zinc |
Oral
|
1 mg/kg/d |
|
Magnesium |
Oral Intravenous |
1-2 mEq/kg/d 0.3-0.5 mEq/ kg over 3 hours
of 50% solution |
|
Elemental iron |
Oral |
5-6 mg/kg/d |
Adapted from Ref. 39
*Doses are provided as a guide only and will need to be adjusted
based on response and levels of the vitamins. #Careful
monitoring required as Vitamin A itself is hepatotoxic; $Vitamin
K1 preparation preferred, as it is safe for G6PD deficient
individuals; **Calcium should always be supplemented along with
Vitamin D. |
Specific treatment
Special infant formula and diets are recommended for
children with specific diagnosis (galactosemia, fructosemia and
tyrosinemia). However these formulae are currently not available in
India. The group feels that steps must be taken to make them available
at the earliest. Treatment with nitisinone (1 mg/kg/d) in addition to
dietary restriction leads to rapid reduction of toxic metabolites in
tyrosinemia. Specific therapy is recommended for patients with CMV
(associated neurological involvement), herpes and toxoplasmosis related
cholestasis. There is no role for steroids in idiopathic neonatal
hepatitis.
In infants with pruritus due to severe cholestasis,
the group recommended, in the following order: Ursodeoxycholic acid
(UDCA) (20 mg/kg/d), rifampicin (5-10 mg/kg/d), and phenobarbitone (5–10
mg/kg/d). Symptom chart should be made for pruritus. Depending on
severity and response to previous agent, add-on drug can be considered.
Appropriate antibiotics depending on the site of infection and culture
sensitivity reports need to be administered in patients with bacterial
sepsis.
Kasai’s PE consists of removal of the atretic
extra-hepatic tissue and a Roux-en-Y jejunal loop anastomosis to the
hepatic hilum. PE may be considered successful if serum bilirubin
normalizes after surgery. In general, over half the patients normalize
their bilirubin after Kasai’s PE if performed within six months [36].
About 20% of all patients undergoing Kasai’s PE during infancy survive
into adulthood with their native liver [36,37]. In children with
progressive familial intrahepatic cholestasis (PFIC) without
decompensated cirrhosis, external and internal biliary diversion has
been shown to be of benefit [38]. Surgery gives excellent results for
choledochal cysts and should be performed as soon as the diagnosis is
made. The recent consensus statement of the Pediatric Gastroenterology
chapter on Acute liver failure gives more details on the management of
liver failure in NC [39].
Liver transplantation
Liver Transplantation, the standard therapy for
decompensated cirrhosis due to any cause, is now well established in
India as well [40]. Any baby, who has had Kasai’s PE and the bilirubin
remains >6 mg/dL, three months after surgery, should be referred to a
transplant center. Babies with BA who present with decompensated
cirrhosis (low albumin, prolonged INR, ascites) are not likely to
improve with a Kasai PE and should be referred for liver
transplantation. Of the 355 transplants in children that have been
performed in India till 2012, 30% have been for BA [41]. Living related
liver transplantation (the vast majority of liver transplants in India
are living related), performed at experienced centers, is associated
with favorable outcomes, with 5- and 10-year survival rates of 98% and
90%, respectively [42-44].
Conclusions and Recommendations
• NC constitutes almost one-third of children
with chronic liver disease in major hospitals in India. BA, NH and
metabolic causes are the most important causes in India.
• Early identification of the cause is essential
for a favorable outcome. This requires specific biochemical tests,
imaging studies and interpretation of histopathology by experienced
personnel and is now possible in major centers all over India.
• The overall outcome in India is far from
satisfactory, due to late referral. The mean age at presentation in
tertiary care centers is still over 3 months compared to recommended
age of less than 60 days.
• To ensure early referral, there is an urgent
need to sensitize pediatricians, obstetricians and other
primary-care physicians on the need for early evaluation. ‘Yellow
alert’ should be extended to an All-India level and the stool color
card should be incorporated in the well-baby cards of IAP and the
Government of India.
• Metabolic diseases (e.g. galactosemia,
fructosemia, hemochromatosis, tyrosinemia) and inherited diseases
like PFIC are increasingly being diagnosed in tertiary centers.
Establishment of regional referral labs will enable greater
diagnosis of causes of NC.
• Ultrasound, isotope scan and liver biopsy
interpretation in experienced hands are effective in ruling out
surgical causes in a majority of cases. If BA cannot be ruled out
with certainty, an experienced surgeon should perform a laparotomy
andintra-operative cholangiography.
• Malnutrition adversely affects the outcome in
infants with cholestasis. Nutritional support and vitamin/mineral
supplementation is recommended in all babies with NC. Special
formulae may have a role in select cases.
• When NC leads to liver failure, LT should be
offered. Success rates in India are comparable to those in the West.
Contributors: VB composed the first draft of this
statement, which after input from AB, JM, YW and AS was circulated to
all the participants.
Funding: Rakhi and Adish Oswal funded the Round
Table Conference in memory of their son Kunwar Viren Oswal.
Competing interests: None stated
Appendix
List of Invited Participants
AK Patwari, Akshay Kapoor, Akshay Saxena, Alok Hemal,
Anjali Kulkarni, Anshu Srivastava, Anupam Sibal (AS), Ashish Bavdekar
(AB), BD Dwivedi, BR Thapa, Dhanasekhar K., John Mathai (JM), Lalit
Bharadia, Malathi Sathiyasekharan, Manoja Das, Narendra K Arora, Neelam
Mohan*, Nishant Wadhwa, Pankaj Vohra, Pawan Rawal*, Praveen Kumar,
Rajeev Tomar, Rajiv Redkar, Rakesh Mishra, SK Mittal*, S Srinivas,
Sarath Gopalan, Seema Alam*, Shrish Bhatnagar*, Subash Gupta, Sujit
Chowdhary*, Sumathi B., Sutapa Ganguly, Ujjal Poddar*, VS
Sankaranarayanan, Veena Malhotra, Vibhor Borkar, Vidyut Bhatia (VB),
Vishnu Biradar, Yogesh Waikar (YW)
*Contributed but could not attend the Round Table
Conference on 8th December 2012.
References
1. Consensus report on neonatal cholestasis syndrome.
Pediatric Gastroenterology Subspecialty Chapter of Indian Academy of
Pediatrics. Indian Pediatr. 2000;37:845-51.
2. Yachha SK, Sharma A. Neonatal cholestasis in
India. Indian Pediatr. 2005;42:491-2.
3. Yachha SK. Cholestatic jaundice during infancy.
Indian J Gastroenterol. 2005;24:47-8.
4. Davis AR, Rosenthal P, Escobar GJ, Newman TB.
Interpreting conjugated bilirubin levels in newborns. J Pediatr.
2011;158:562-5.
5. Moyer V, Freese DK, Whitington PF, Olson AD,
Brewer F, Colletti RB, et al. Guideline for the evaluation of
cholestatic jaundice in infants: recommendations of the North American
Society for Pediatric Gastroenterology, Hepatology and Nutrition. J
Pediatr Gastroenterol Nutr. 2004;39:115-28.
6. Yachha SK, Khanduri A, Kumar M, Sikora SS, Saxena
R, Gupta RK, et al. Neonatal cholestasis syndrome: an appraisal
at a tertiary center. Indian Pediatr. 1996;33:729-34.
7. Arora NK, Arora S, Ahuja A, Mathur P, Maheshwari
M, Das MK, et al. Alpha 1 antitrypsin deficiency in children with
chronic liver disease in North India. Indian Pediatr. 2010;47:1015-23.
8. Yachha SK, Sharma BC, Khanduri A, Srivastava A.
Current spectrum of hepatobiliary disorders in northern India. Indian
Pediatr. 1997;34:885-90.
9. Alagille D, Habib EC, Thomassin N. L’atresie des
voies biliaires extrahepatiques permeables chez l’enfant. J Par Pediatr
1969:301-18.
10. Kamath BM, Spinner NB, Piccoli DA. Alagille
Syndrome. In: Suchy F, Sokol RJ, Balistreri WF, editors. Liver Disease
in Children. Third ed. New York: Cambridge University Press; 2007. p.
326-45.
11. Mizuta K, Sanada Y, Wakiya T, Urahashi T, Umehara
M, Egami S, et al. Living-donor liver transplantation in 126
patients with biliary atresia: single-center experience. Transplant
Proc. 2010;42:4127-31.
12. Sanghai SR, Shah I, Bhatnagar S, Murthy A.
Incidence and prognostic factors associated with biliary atresia in
Western India. Ann Hepatol. 2009;8:120-2.
13. Ahmad M, Jan M, Ali W, Shabir ud d, Bashir C,
Iqbal Q, et al. Neonatal cholestasis in Kashmiri children. JK
Pract. 2000;7:125-6.
14. Khalil S, Shah D, Faridi MM, Kumar A, Mishra K.
Prevalence and outcome of hepatobiliary dysfunction in neonatal
septicaemia. J Pediatr Gastroenterol Nutr. 2012;54:218-22.
15. Poddar U, Thapa BR, Das A, Bhattacharya A, Rao
KL, Singh K. Neonatal cholestasis: differentiation of biliary atresia
from neonatal hepatitis in a developing country. Acta Paediatr.
2009;98:1260-4.
16. Rastogi A, Krishnani N, Yachha SK, Khanna V,
Poddar U, Lal R. Histopathological features and accuracy for diagnosing
biliary atresia by prelaparotomy liver biopsy in developing countries. J
Gastroenterol Hepatol. 2009;24:97-102.
17. Shah I, Bhatnagar S. Clinical profile of chronic
hepatobiliary disorders in children: experience from tertiary referral
centre in Western India. Trop Gastroenterol. 2010;31:108-10.
18. Yachha SK, Mohindra S. Neonatal cholestasis
syndrome: Indian scene. Ind J Pediatr. 1999;66:S94-6.
19. Chen SM, Chang MH, Du JC, Lin CC, Chen AC, Lee
HC, et al. Screening for biliary atresia by infant stool color
card in Taiwan. Pediatrics. 2006;117:1147-54.
20. Burton BK. Inborn errors of metabolism in
infancy: a guide to diagnosis. Pediatrics. 1998;102:E69.
21. Lien TH, Chang MH, Wu JF, Chen HL, Lee HC, Chen
AC, et al. Effects of the infant stool color card screening
program on 5-year outcome of biliary atresia in Taiwan. Hepatology.
2011;53:202-8.
22. Lally KP, Kanegaye J, Matsumura M, Rosenthal P,
Sinatra F, Atkinson JB. Perioperative factors affecting the outcome
following repair of biliary atresia. Pediatrics. 1989;83:723-6.
23. Serinet MO, Wildhaber BE, Broue P, Lachaux A,
Sarles J, Jacquemin E, et al. Impact of age at Kasai operation on
its results in late childhood and adolescence: a rational basis for
biliary atresia screening. Pediatrics. 2009;123:1280-6.
24. Lampela H, Ritvanen A, Kosola S, Koivusalo A,
Rintala R, Jalanko H, et al. National centralization of biliary
atresia care to an assigned multidisciplinary team provides high-quality
outcomes. Scand J Gastroenterol. 2012;47:99-107.
25. McKiernan PJ, Baker AJ, Kelly DA. The frequency
and outcome of biliary atresia in the UK and Ireland. Lancet.
2000;355:25-9.
26. Whitington PF, Freese DK, Alonso EM,
Schwarzenberg SJ, Sharp HL. Clinical and biochemical findings in
progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol
Nutr. 1994;18:134-41.
27. Hsiao CH, Chang MH, Chen HL, Lee HC, Wu TC, Lin
CC, et al. Universal screening for biliary atresia using an
infant stool color card in Taiwan. Hepatology. 2008;47:1233-40.
28. Niedbala A, Lankford A, Boswell WC, Rittmeyer C.
Spontaneous perforation of the bile duct. Am Surg. 2000;66:1061-3.
29. Lee WS, Looi LM. Usefulness of a scoring system
in the interpretation of histology in neonatal cholestasis. World J
Gastroenterol. 2009;15:5326-33.
30. Singh R, Thapa BR, Kaur G, Prasad R. Biochemical
and molecular characterization of GALT gene from Indian galactosemia
patients: identification of 10 novel mutations and their structural and
functional implications. Clin Chim Acta. 2012;414:191-6.
31. Singh R, Kaur G, Thapa BR, Prasad R, Kulkarni K.
A case of classical galactosemia: identification and characterization of
3 distinct mutations in galactose-1-phosphate uridyl transferase (GALT)
gene in a single family. Indian J Pediatr. 2011;78:874-6.
32. Bijarnia S, Puri RD, Ruel J, Gray GF, Jenkinson
L, Verma IC. Tyrosinemia type I—diagnostic issues and prenatal
diagnosis. Indian J Pediatr. 2006;73:163-5.
33. Suchy FJ. Neonatal cholestasis. Pediatr Rev.
2004;25:388-96.
34. Feranchak AP, Sokol RJ. Medical and nutritional
mManagement of cholestasis in infants and children. In: Suchy FJ, Sokal
RJ, Balistreri WF, editors. Liver Diseases in Children. 3rd ed. New
York: Cambridge University Press; 2007. p. 190-231.
35. Venigalla S, Gourley GR. Neonatal cholestasis.
Semin Perinatol. 2004;28:348-55.
36. Sokol RJ, Shepherd RW, Superina R, Bezerra JA,
Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia:
summary of a National Institutes of Health workshop. Hepatology.
2007;46:566-81.
37. Bassett MD, Murray KF. Biliary atresia: recent
progress. J Clin Gastroenterol. 2008;42:720-9.
38. Sharma D, Shah UH, Sibal A, Chowdhary SK.
Cholecystoappendicostomy for progressive familial intrahepatic
cholestasis. Indian Pediatr. 2010;47:626-8.
39. Bhatia V, Bavdekar A, Yachha SK. Management of
acute liver failure in infants and children: Consensus statement of the
Pediatric Gastroenterology Chapter, Indian Academy of Pediatrics. Indian
Pediatr. 2013;50:477-82.
40. Poonacha P, Sibal A, Soin AS, Rajashekar MR,
Rajakumari DV. India’s first successful pediatric liver transplant.
Indian Pediatr. 2001;38:287-91.
41. Sibal A. Pediatric Liver transplantation in
India: Past, Present and future. 21th Annual Conference of Indian
National Association for Study of the Liver [INASL] 2013; Hyderabad,
India.
42. Utterson EC, Shepherd RW, Sokol RJ, Bucuvalas J,
Magee JC, McDiarmid SV, et al. Biliary Atresia: Clinical
Profiles, Risk Factors, and Outcomes of 755 Patients Listed for Liver
Transplantation. J Pediatr. 2005;147:180-5.
43. Wang SH, Chen CL, Concejero A, Wang CC, Lin CC,
Liu YW, et al. Living donor liver transplantation for biliary
atresia. Chang Gung Med J. 2007;30:103-8.
44. Kaur S, Wadhwa N, Sibal A, Jerath N, Sasturkar S. Outcome of live
donor liver transplantation in Indian children with bodyweight 7.5 kg.
Indian Pediatr. 2011;48:51-4.
|
|
|
 |
|
