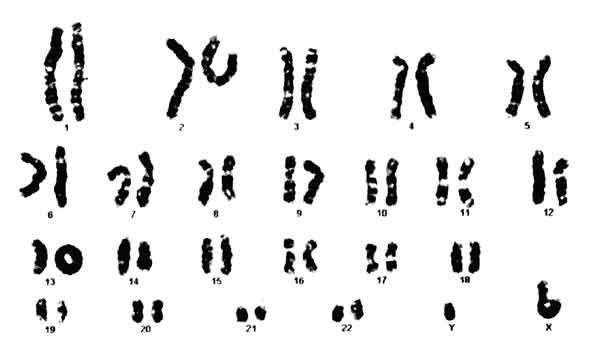|
|
Case Reports Indian Pediatrics 2006; 43:258-260 |
||||||||
|
Ring Chromosome 13 in an Infant with Ambiguous Genitalia |
||||||||
|
From the Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226 014, UP, India. Correspondence to: Dr. Shubha R. Phadke, Associate Professor, Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226 014, UP. India. E-mail: [email protected] Manuscript received: May 20, 2005; Initial review
completed: June 14, 2005;
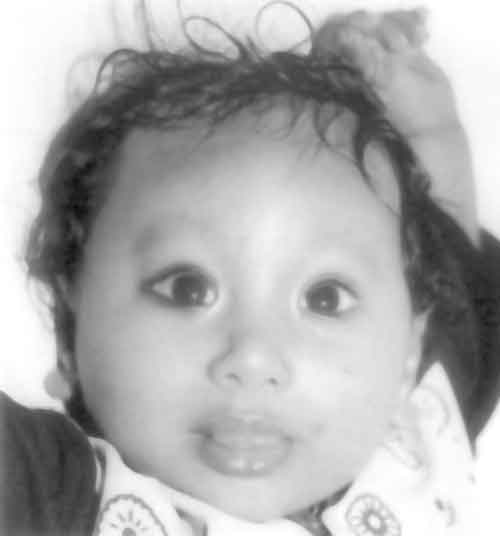
Lejune, et al.(1) described ring chromo-some 13 in 1968. The classic mode of formation of a ring chromosome is breakage of the ends of both arms of a chromosome with fusion of the broken ends resulting in the loss of the distal fragments(2). The clinical phenotype will depend on loss of genetic material. Phenotype similar to that of large deletion of 13q has been described in cases with "ring 13 chromosome syndrome". We describe a typical case of ring 13 syndrome along with the review of literature(1). Case Report The present case was a 4-month-old baby with developmental delay. He had ambiguous genitalia and was reared as a male. The baby was the third child of non-consanguineous parents. He was born of a full term normal vaginal delivery. There was no history of any significant antenatal problems, drug exposure or medical illness in the mother. The parents noticed the ambiguous genitalia soon after birth and had difficulty in assigning sex. He did not have any neonatal problems like vomiting, seizures or salt wasting crisis. Parents noticed developmental delay at the age of four months. The elder siblings are healthy. There is no family history of perinatal or neonatal death, infertility or ambiguity of sex. On examination the weight (4.5 kg), length (56 cm) and occipitofrontal circumference (38 cm) were less than –3 SD for Indian standards. The body proportion was normal for the age (1.6 : 1). Baby had microcephaly, sloping forehead, prominent metopic sutures, hypertelorism, epicanthic fold, mongoloid slant, anteverted nares and small pointed chin (Fig. 1). Phallus was very small (1 cm) embedded in the bifid scrotal folds and gonads were palpable in the scrotal folds bilaterally (Fig. 2). There was a single urogenital opening in the perineum, below and left to the phallus. There was no hyper pigmentation of genitalia. He did not have skeletal malformation except clinodactyly on both sides. Baby had developmental delay, hypertonia and exaggerated deep tendon reflexes. He did not have hepatosplenomegaly and heart was clinically normal. Ultrasonogram of the abdomen and magnetic resonance imaging (MRI) of pelvis did not reveal any mullarian structures or any other abnormalities. Hematological investigations, liver function and renal function tests were normal.
Metaphase chromosome preparation from the proband was made from lymphocyte culture and stained with Giemsa (GTG banding). A total of 20 metaphases were analyzed. Cytogenetic analysis showed 46 chromosomes including a ring chromosome 13 in all metaphases, one X and one Y chromosome [46, XY, r(13) (p11q34)] (Fig. 3). Karyotype of the parents did not reveal ring chromosome or any other abnormality.
Discussion We describe a baby with ring chromosome 13. His features namely microcephaly, developmental delay, growth retardation, dysmorphic features and genital abnormalities are classical presentations of a ring chromosome 13 syndrome. Other features reported in ring chromosome 13 syndrome are skeletal malformation (hand, foot and toe abnormalities) and cerebral malformations. Many cases with partial deletion of long arm of chromosome 13 are described in the literature. These cases include terminal or interstitial deletion, deletion due to un-balanced translocation or ring chromosomes. The phenotype is variable depending on the length of deleted segment and breakpoints. There is no definite phenotypic correlation with extent of deletions. Microcephaly, facial dysmorphism, ambiguous genitalia, growth and developmental delay present in this case are reported with ring chromosome 13 and deletion of 13q(3). The severe malformation like encephalocoele, anencephaly and atelencephaly are also reported with ring chromosome 13(4). Nine cases of ring chromosome 13 with breakpoint at q34 are described in the literature. Two of them had ambiguous genitalia similar to our case(5). Out of 34 cases of ring 13 with proximal breakpoints on 13q and involving band q34 in the deleted segment, 14 cases had ambiguous genitalia. Two cases with small terminal deletions involving q34 are reported to have penoscrotal inversion and hypospadias(6). On the other hand, out of 9 cases with interstitial deletion of 13q where q34 was not deleted, none had genital abnormality. Band q34 is always deleted in cases of ambiguous genitalia with chromosome 13 abnormalities. This suggests that the 13q34 has genes involved in genital development. Ring 13 is usually not inherited as the phenotype is too severe to reproduce. Hence, the risk of recurrence in the siblings of a child with ring 13 is negligible to 1%. Recently, a case of transmission of ring 13 from mother to daughter is reported. Ring chromosome has a small subtelomeric deletion of 13q detectable by fluorescence in situ hybridization only and the mother had borderline mental retardation(7). Ambiguous genitalia have also been reported with chromosomal anomalies of other autosomes like trisomy 13 and trisomy 18. Hence, analysis of chromosomes should not be limited to presence or absence of ‘Y’ only; especially if other features like dysmorphism and developmental delay are present. Funding: None. Competing interests: None.
| ||||||||
|
References | ||||||||
| ||||||||
![]()