|
|
|
Indian Pediatr 2013;50: 591-593 |
 |
Clinical and Molecular Characterization of
Patients with Gross Hypotonia and Impaired Lower Motor Neuron
Function
|
|
Jayesh Sheth, Harsh Patel, *Sanjiv Mehta, Stuti Tewari,
and Frenny Sheth
From FRIGE ’s Institute of Human Genetics,
FRIGE House, Jodhpur Gam Road, Satellite, Ahmedabad and *Usha-Deep
Children Neurology and Epilepsy Clinic, Mansarovar Complex, Naranpura,
Ahmedabad, Gujarat, India.
Correspondence to: Dr Jayesh J Sheth, Institute of
Human Genetics, FRIGE FRIGE ’s House, Jodhpur Gam Road, Satellite,
Ahmedabad 380 015, India.
Email: [email protected]
Received: September 28, 2012;
Initial review: October 09, 2012;
Accepted: October 25, 2012.
PII: S097475591200843
|
Spinal muscular atrophy (SMA) represents the second most common
fatal autosomal recessive disorder after cystic fibrosis. Due to the
high carrier frequency, the burden of this genetic disorder is very
heavy in developing countries like India. The aim was to study the
clinical and molecular characteristics of patients suspected with
SMA. It was a cross sectional study of 105 cases from January 2008
to August 2012. Patients’ demographic and presenting features and
PCR findings were noted. 65 (62%) cases had a confirmed diagnosis of
SMA. Werdnig Hoffman disease (SMA type I) was the commonest variant
seen in 34 (52.3%) children. Molecular analysis demonstrated
deletion of both exon 7 and 8 of SMN1 gene in 83.1% of cases.
Key words: Hypotonia, India, Spinal Muscular Atrophy,
SMN1, SMN2.
|
|
Spinal muscular atrophy (SMA) is the most common
genetic cause of infant mortality [1] with an incidence of 1 in
6,000-10,000 live births [2]; and a carrier frequency of 1:50 [3]. It
results from homozygous deletions of exon 7 and 8 involving the SMN1
gene located on chromosome 5q13 [4]. Homozygous SMN2 detection,
although found in 5-9% of normal control, may be associated with disease
phenotype in selected cases [2,4,5]. Most cases of SMA have autosomal
recessive inheritance; however, autosomal dominant and X-linked
inheritance has also been documented [6].
SMA phenotype varies, depending on the age of onset
and motor development milestones [7]. Since in India, SMA remains highly
under-diagnosed, present study aimed to analyse the clinical
characterization of patients with gross hypotonia and impaired lower
motor neuron function and their further molecular confirmation by SMN
gene study.
Methods
This is a cross-sectional study carried out on
patients referred between the period of January 2008 to August 2012 from
Gujarat and its vicinity. The main clinical phenotype were marked
hypotonia, diffuse proximal muscle weakness, tongue fasciculation along
with absent or greatly decreased deep tendon reflexes and
electromyographic (EMG) evidence of denervation on systemic examination.
After approval from the ethical committee, 105 subjects were included in
the study and their name, age, gender, other demographic findings,
presenting signs and symptoms, family history were noted. Molecular
analysis was carried out after taking informed written consent.
Gene analysis: Genomic DNA was isolated from
blood samples using salting out method [8]. Deletion of exon 7 and 8 of
SMN1 and SMN2 gene was carried out by polymerase chain
reaction (PCR) amplification and restriction endonuclease digestion. PCR
products of exon 7 were digested with Dra I, and exon 8 with Dde I and
run on 2.5% agarose gel electrophoresis at 100v and observed under UV
transilluminator.
Results
Of 105 subjects presenting with suspicion of SMA,
65(62%) were consistent with the clinical diagnosis of SMA. Out of
these, 34(52.3%) children were in the acute infantile group with onset
within 6 months of age, 12 (18.5%) were in the range of 7-18 months
falling in the category of chronic childhood, 18 (27.7%) subjects
belonged to the chronic juvenile category while only 1 subject (1.5%)
was more than 30 years of age at the time of presentation as shown in
Table I. Males had a greater preponderance than females in
our study in the ratio of 1.5:1. This skewed ratio in favour of males
was most striking in SMA type I subjects.
TABLE I Summary of Confirmed Cases of SMA
|
Type of SMA |
Most common presentation |
Number of cases |
|
Type I |
Hypotonia and decreased |
34 (22 males)
|
|
limb movement |
|
|
Type II |
Delayed motor milestones |
12 (7 males)
|
|
and hypotonia |
|
|
Type III |
Proximal muscle weakness |
18 (9 males) |
|
Type IV |
Weakness in lower limbs |
1 male |
Molecular study demonstrated that, 83.1% of cases
showed deletion of both the exons 7 and 8 of SMN1 gene while 6.2%
and 4.6% cases showed deletion of only exon 7 and exon 8, respectively.
Exon 7 and/or 8 deletion in SMN2 gene was observed in 4.6% of
cases. Deletion of both exons of SMN1 and SMN2 gene was
detected in one case. Out of 12 cases with SMA type II, none showed
deletion in exon 7 and/or 8 of the SMN2 gene while the only case
of adult onset SMA (type IV) had deletion in exon 7 of SMN2 gene
(Fig. 1).
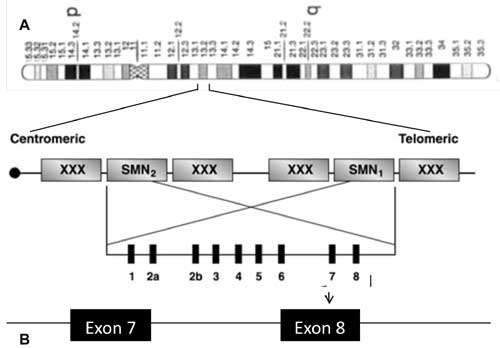 |
|
Fig. 1 Schematic representation of SMA
region on chromosome 5 and localization of SMN1 (telomeric copy)
and SMN 2 (centromeric copy) on 5q13.
|
Discussion
In our study majority of patients presented within
the first 6 months and were of SMA type I (53.3%) which is similar to
the observations in other studies [6.9]. Males had a greater
preponderance which is in concordance with earlier study [6] while a
female preponderance was reported in an Egyptian cohort [9].
Different genes and microsatellite markers have been
identified in the 5q region that can be deleted in SMA patients.
Homozygous deletions of the SMN1 gene are detected in more than
90% of the SMA type I to III patients and only exceptionally in SMA type
IV [10]. The number of SMN2 copies correlates with the SMA
subtype, age of onset, and length of survival [11]. It has been observed
that 95% of SMA type I patients have only 1-2 copies of SMN2,
whereas almost all patients belonging to type III had 3 or more copies
and a less severe disease course [12]. The most common deletion found in
our study was of SMN1 gene with deletion of both exons 7 and 8 in
majority of cases (83.1%). Similar observation was made in the cohort of
Pakistani and Egyptian children [6,9]. Deletions of either exon 7 or
exon 8 of SMN1 gene and SMN2 gene were observed only in
around 5% of cases. Other studies have reported exon 7 deletion of
SMN1 gene in 18.2% of cases while none of the cases had deletion of
only exon 8 [9] and homozygous deletion of SMN2 gene in a
childhood onset SMA [13]. A study in Korean population demonstrated an
association between sporadic motor neuron disease and SMN2
deletion in adults [14]. With increasing identification of the
underlying genetic defects, clinical spectrum and presentation, the
awareness of the disease has improved. However, because no cure is yet
available, genetic counselling and prognostic considerations are of
great importance. With the availability of genetic testing, it is now
possible to diagnose these children early so that appropriate
counselling can be offered to the family on the risk of future
pregnancies. Early identification by genetic testing has also made early
antenatal diagnosis possible. Thus spreading awareness about this lethal
but preventive disease becomes imperative.
Acknowledgments: The referring clinicians for
their cooperation and support.
Funding: None; Competing interests: None
Stated.
|
What This Study Adds?
•
Patients presenting with
hypotonia and clinical features of lower motor neuron disease
carry high level of clinical suspicion for SMA and need further
confirmation.
|
References
1. Markowitz JA, Singh P, Darras BT. Spinal
muscular atrophy: a clinical and research update. Pediatr
Neurol. 2012;46:1-12.
2. Panigrahi I, Kesanri A, Phadke SR, Mittal B.
Clinical and molecular diagnosis of spinal muscular atrophy. Neurol
India. 2002;50:117-22.
3. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet.
2008;371:2120-33.
4. Lefebvre S, Burglen L, Reboullet S, Clermont O,
Burlet P, Viollet L, et al. Identification and characterization
of a spinal muscular atrophy-determining gene. Cell. 1995;80:155-65.
5. Gerard B, Ginet N, Matthijs G, Evrard P, Baumann
C, Da Silva F, et al. Genotype determination at the survival
motor neuron locus in a normal population and SMA carriers using
competitive PCR and primer extension. Hum Mutat. 2000;16:253-63.
6. Ibrahim S, Moatter T, Saleem AF. Spinal muscular
atrophy: Clinical spectrum and genetic mutations in Pakistani children.
Neurol India. 2012;60:294-8.
7. Munsat TL Workshop report. International SMA
collaboration. Neuromusc Disord. 1991;1:81.
8. Miller SA, Dykes DD, Polesky HF. A simple salting
out procedure for extracting DNA from human nucleated cells. Nucleic
Acids Res. 1988;16:1215.
9. Shawky RM, El-Syed NS. Clinico-epidemiologic
characteristics of spinal muscular atrophy among Egyptians. Egypt J Med
Hum Genet. 2011;12:25-30.
10. Klaus Zerres, Sabine Rudnik-Schoneborn. Spinal
Muscular Atrophies. In: David L. Rimoin, J. Michael Connor, Reed
E. Pyeritz, Bruce R. Korf, Editors. Principles and Practice of Medical
Genetics. 5th ed. Elsevier press; 2002. pp.3001-23.
11. Feldkotter M, Schwarzer V, Wirth R, TF Weinker
and Brunhilde Wirth. Quantitative analyses of SMN1 and SMN2 based on
real-time LightCycler PCR: fast and highly reliable carrier testing and
prediction of severity of spinal muscular atrophy. Am J Hum Genet.
2002;70: 358-68.
12. Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra
MS, Wirth B, et al. Molecular analysis of spinal muscular atrophy
and modification of the phenotype by SMN2. Genet Med. 2002;4:20-6.
13. Srivastava S, Mukherjee M, Panigrahi I, Pandey
SG, Pradhan S, Mittal B. SMN2-deletion in childhood-onset spinal
muscular atrophy. Am J Med Genet. 2001;101:198-202.
14. Kim J, Lee SG, Choi YS, Kang SW, Lee JB, Choi JR,
et al. Association between Survivor Motor Neuron 2 (SMN2) gene
homozygous deletion and sporadic lower motor neuron disease in a Korean
population. Ann Clin Lab Sci. 2010;40:368-74
|
|
|
 |
|
