|
|
|
Indian Pediatr 2009;46: 525-527 |
 |
Chronic Eosinophilic Leukemia With a Unique
Translocation |
|
RS Arora
From the Department of Pediatrics, Moolchand Khairatiram
Hospital, New Delhi, India.
Correspondence to: Dr RS Arora, 19 Wet Earth Green,
Swinton, Manchester, M27 8AL.
E-mail:
[email protected]
Manuscript received: February 18, 2008;
Initial review: May 12, 2008;
Accepted: May 30, 2008.
|
|
Abstract
We report a case of chronic eosinophilic leukemia in
a 9 year old girl who presented with anemia, thrombocytopenia,
leucocytosis (mostly dysplastic eosinophils), lymphadenopathy and
hepatosplenomegaly. There was no increase in blasts but myelofibrosis
was seen in the bone marrow. A previously unreported translocation
46,XX,t(1;4)(q24;q35), was found on cytogenetic analysis and involvement
of the myocardium was also present. Shortly after commencing steroids,
the family abandoned therapy.
Keywords: Cardiomyopathy, Chronic eosinophilic leukemia,
Hypereosinophilia, t(1;4)(q24;q35)
|
|
Eosinophilia
in children is usually due to allergic rhinitis, asthma, and atopic
dermatitis. Infrequent causes include Churg–Strauss vasculitis, hyper-IgE
syndrome, tropical pulmonary eosinophilia, eosinophilic gastroenteritis
and connective tissue disorders. There are also a diverse group of
myeloproliferative and neoplastic diseases such as acute and chronic
eosinophilic leukemia, chronic granulocytic leukemia, and acute myeloid
and lymphoblastic leukemia. When no etiology is established, it is termed
as idiopathic hypereosino-philic syndrome (IHES) as described by Chusid,
et al. in 1975(1). Sustained hypereosinophilia, whether reactive,
clonal or idiopathic could potentially lead to eosinophilic end organ
damage. The frequency of organ involvement in a review of 105 patients was
hematologic 100%, cardiovascular 58%, cutaneous 56%, neurologic 54%,
pulmonary 49%, splenic 43%, hepatic 30% and ocular 23%(2).
Case Report
A nine year old girl presented to Moolchand Khairatiram
Hospital in New Delhi with a one month history of fever and bone pains.
There was no history of bleeding from any site, allergy to drugs, history
of asthma or worm infestation. There was no contact with tuberculosis. On
examination the child was pale and there were no petechiae. There was
significant bilateral axillary lymphadenopathy, hepatospleno-megaly and
sternal tenderness. Systemic exami-nation was otherwise unremarkable.
Her initial blood count revealed hemoglobin of 8.0 g/dL,
marked eosinophilia (total leukocyte count of 162×10 3/µL
with absolute eosinophil count of 140×103/µL) and platelet count of
102×103/µL. The blood film confirmed the marked eosinophilia with
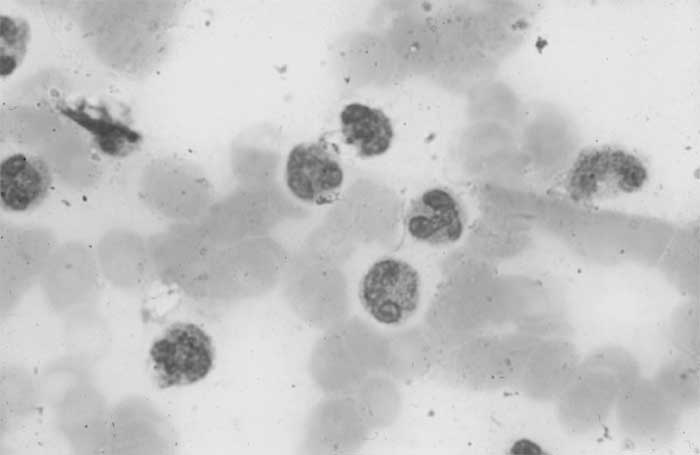
abnormally lobulated and hypogranular forms (Fig.1).
Eosinophil metamyelocytes and myelocytes were present but no blasts were
seen. Erythrocyte sedimentation rate (ESR) was 32mm/hour, liver enzymes
were slightly above the normal range and renal function tests were normal.
The immunoglobulin E (IgE) level was 173 IU/mL (normal <180 IU/mL). Urine
and stool analysis was normal. Bone marrow showed increased cellularity
with near complete population of eosinophils and eosinophil precursors but
no increase in blasts. Megakaryocytes were seen and erythroid series was
normal. Myelofibrosis was also seen. Karyotyping revealed presence of
translocation 46,XX,t(1;4)(q24;q35). On echocardiogram the apex of both
left and right ventricles appeared obliterated by echogenic tissue. The
ejection fraction was 60%.
 |
|
Fig.1 Blood smear showing abnormal, mature eosinophils,
abnormally lobulated with unilobed and trilobed cells. There is
marked hypogranularity.
|
The final diagnosis was chronic eosinophilic leukemia
with eosinophilic cardiomyopathy. The child was started on steroids to
minimize organ damage from the eosinophilic granules. Unfortu-nately,
before further treatment could be commenced, the family self-discharged
themselves and failed to follow-up. Attempts were made to contact the
family by phone and post with no success.
Discussion
Eosinophilia can be classified into mild (eosinophils
<1.5×10 3/µL), moderate (eosinophils
1.5-5×103/µL) or severe (>5×103/µL)(3). The increase in eosinophil count
in most cases is because of generation of cytokines, particularly GM-CSF,
IL-3 and IL-5 which stimulate its production and differentiation. It is
important to distinguish between reactive, clonal and idiopathic
eosinophilia as their treatment and prognoses are different. The most
easily available methods being bone marrow cytogenetic analysis and
fluorescent in-situ hybridization (FISH). The detection of any
abnormalities confirms a clonal disorder(4). Indirectly, the presence of
dysplastic eosinophils, increased serum B12, increased serum tryptase,
anemia/thrombocytopenia, increased bone marrow cellularity with left
shift, myelofibrosis, and dysplastic mast cells or megakaryocytes in bone
marrow also favors diagnosis of clonal eosino-philia(3,5). The absence of
increased blasts, increased mast cells and negative Philadelphia and
BCR-ABL probes on cytogenetic analysis suggests chronic eosinophilic
leukemia.
Our child had a karyotype of 46,XX,t (1;4)(q24;q35).
This translocation has not been reported previously. However, a balanced
trans-location in all cells with unusual breakpoints could also have been
possible. As the patient abandoned further management we were unable to
either confirm this or do the mapping of the genes at the breakpoints to
confirm specific gene involvement and possible fusion gene formation.
Several cytogenetic abnormalities have been reported(6,7), including
trisomies of chromosome 1,8,10 and 15, monosomy of 7 and translocations of
the long arm of chromosome 5 q31-33 zone (where the genes encoding for
IL-5, GM-CSF, and IL-3 are localized). The new advancement has been the
discovery of the FIP1L1-PDGFRA (F-P) fusion gene created by the
del(4)(q12q12), an 800-kb deletion on chromosome 4q12 and the excellent
response to imatinib of this subgroup of F-P + chronic eosinophilic
leukemia (CEL) patients. Based on the limited number of patients
evaluated, this group currently accounts for 50% to 60% of all HES and CEL
cases(8). Similar success to imatinib has also been seen in patients with
chronic myeloproliferative disease and eosinophilia where activation of
the gene for platelet-derived growth factor receptor beta (PDGFRB) was
caused by a t(5;12)(q33;p13) translocation(9).
This distinction is important because there is a
potentially curative treatment available for clonal marrow disorders
(particularly F-P+ hypereosinophilia). For idiopathic HES the aim is to
limit eosinophilic end organ damage with use of steroids as first-line
therapy. In the past hydroxyurea has been used in those resistant to
steroids but now interferon-alpha is considered the treatment of choice in
corti-costeroid-refractory patients(10). If the CEL is F-P+ then imatinib
is the drug of choice(11). Also, since clonal cytogenetic abnormalities
may develop during the course of IHES, it is important to make regular
cytogenetic and more sensitive assessment of clonality on bone marrow
samples. In presence of signs of malignant transformation chemotherapy and
bone marrow or stem cell transplantation would be needed.
Acknowledgments
I would like to thank Dr Rob Wynn, Mr Nick Telford and
Dr Oliver Rackham for their useful comments during preparation of this
manuscript.
Funding: None.
Competing interests: None stated.
References
1. Chusid MJ, Dale DC, West BC, Wolff SM. The
hypereosinophilic syndrome: analysis of fourteen cases with review of the
literature. Medicine (Baltimore) 1975; 54: 1-27.
2. Weller PF, Bubley GJ. The idiopathic hyper-eosinophilic
syndrome. Blood 1994; 83: 2759-2779.
3. Brito-Babapulle F. Clonal eosinophilic disorders and
the hypereosinophilic syndrome. Blood Rev 1997; 11: 129-145.
4. Brito-Babapulle F. The eosinophilias, including the
idiopathic hypereosinophilic syndrome. British Hematol 2003;121: 203-223.
5. Klion AD, Robyn JA, Akin C, Noel P, Brown MR, Law
MA, et al. Molecular remission and reversal of myelofibrosis in
response to imatinib mesylate treatment in patients with the
myeloproliferative variant of hypereosinophilic syndrome. Blood
2004; 103: 473-478.
6. Oliver JW, Deol I, Morgana DL, Tonk VS. Chronic
eosinophilic leukemia and hypereosinophilic syndromes. Proposal for
classification, literature review, and report of a case with a unique
chromosomal abnormality. Cancer Genet Cytogenet 1998; 107: 111-117.
7. Bain BJ. Cytogenetic and molecular genetic aspects
of eosinophilic leukaemias. British Haematol 2003; 122: 173-179.
8. Gotlib J, Cools J, Malone JM 3rd, Schrier SL,
Gilliland DG, Coutre SE. The FIP1L1-PDGFR alpha fusion tyrosine kinase in
hypereosinophilic syndrome and chronic eosinophilic leukemia: implications
for diagnosis, classification, and management. Blood 2004; 103:
2879-2891.
9. Apperley JF, Gardembas M, Melo JV, Russell-Jones R,
Bain BJ, Baxter EJ, et al. Response to imatinib mesylate in
patients with chronic myeloproliferative diseases with rearrangements of
the platelet-derived growth factor receptor beta. N Engl J Med 2002; 347:
481-487.
10. Butterfield JH, Gleich GJ. Interferon-alpha
treatment of six patients with the idiopathic hypereosinophilic syndrome.
Ann Intern Med 1994; 121: 648–653.
11. Roufosse F, Cogan E, Goldman M. Recent advances in
pathogenesis and management of hypereosinophilic syndromes. Allergy 2004;
59: 673–689.
|
|
|
 |
|
