|
|
Images in Clinical Practice Indian Pediatrics 2002; 39:594-595 |
|
Mucopolysaccharidosis Types IV and VI |
|
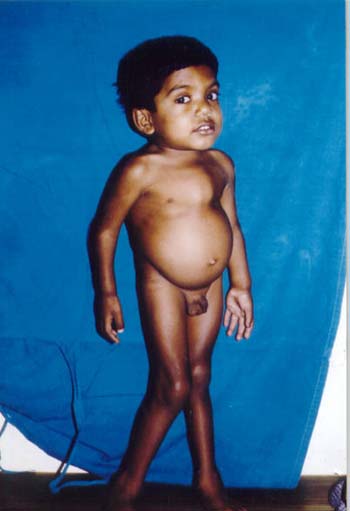
A 6-year-old boy born to consanguineous parents presented with short stature and deformity of chest and limbs. Clinical examination revealed a very short child with the height of 86 cm (height age 2½ years). He had mild coarse facies, clear corneas, short neck and normal intelligence. Other salient features were short trunk, kyphosis, pectus carinatum, protruded abdomen and knock knees (Fig. 1). Liver was palpable 6 cm below the right costal margin and spleen was 2 cm palpable. The blood picture was normal and urine was positive for mucopolysaccharides. X-ray spine showed marked platyspondyly typical of Morquio Syndrome. Echocardio-gram revealed grade II mitral valve prolapse.
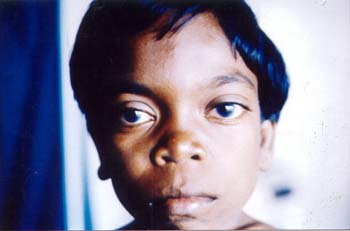
Fig. 1. Photograph showing short neck, short trunk, pectus carinatum and knock knees. A 12-year-old girl born to non-consanguineous parents presented with short stature and difficulty in walking. On examination she was very short with the height of 103 cm (height age 5 years). She had coarse facies, corneal clouding, large mouth, anteverted nose and normal intelligence (Fig.2). Short neck, short trunk, prominent sternum, kyphosis, widened wrist, stubby hands and genu valgum were other prominent features. Liver was palpable 3 cm below the right costal margin but spleen was not palpable. Her urine was positive for mucopolysaccharides. Peripheral blood smear showed Alder-reilly bodies (metachromatic granules) in the neutrophils. X-rays of head, hand, chest and spine showed dysostosis multiplex. Echocardiogram revealed mitral valve prolapse with mitral regurgitation.
Fig. 2. Photograph showing coarse facies, corneal clouding and large mouth. Mucopolysaccharidosis Type-IV is an autosomal recessive disorder, characterized by short stature, severe kyphosis, knock knees and normal intelligence. Cloudy cornea becomes evident by slit lamp examination, usually after 5-10 years of age. A severe form (Type-IV A) and a mild form (Type-IV B) are recognized. The basic defect in Type-IV A is deficiency of N-acetyl galactosamine-6-sulfatase, whereas in Type-IV B there is deficiency of b-galactosidase. Severe defects of vertebrae may result in cord compression or respiratory insufficiency which may lead to death prior to 20 years of age. Mucopolysaccharidosis Type-VI is also an autosomal recessive disorder characterized by coarse facies, cloudy corneas and normal intelligence. Based on the severity mild, moderate and severe subtypes are recognized. Severe subtype is characterized by short stature, short neck and trunk, prominent sternum and genu valgum. Cytoplasmic granules called Alder-reilly bodies are present in the neutrophils. The molecular defect in this syndrome is deficiency of N-acetyl galactosamine-4-sulfatase. The gene for this enzyme maps to chromosome 5q13-q14. A.M. Vijayalakshmi, Associate Professor, Department of Pediatrics, PSG Institute of Medical
Sciences Peelamedu, Coimbatore 641 004, Tamil Nadu, India.
|
![]()