|
|
Case Reports Indian Pediatrics 2000;37: 673-676 |
||||||||||||||
|
Aicardi Syndrome with Dandy-Walker Malformation |
||||||||||||||
|
Aicardi et al. in 1965 described 8 cases with a triad of spasm in flexion, callosal agenesis and ocular abnormalities(1). This clinical syndrome now includes other features like vertebral anomalies, cortical heterotopias, hypotonia, mental subnormality, poor life expectancy, characteristic electrophysiological and radiological abnormalities(2). This disorder has been observed usually in females with the exception of two male infants(3). We report a typical case of Aicardi syndrome with Dandy walker malformation which is an unusual, association.
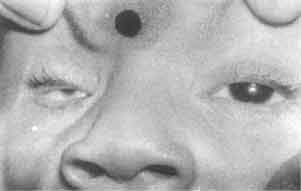
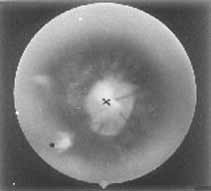
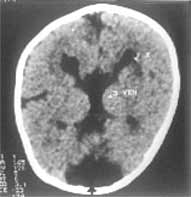
A 7-month-old girl presented to us with infantile spasm since the age of two months. She was born by an uncomplicated normal vaginal delivery at term to a primigravida mother. The parents were non-consanguineous and there was no significant antenatal illness. The symptoms started initially with myoclonic jerks and occasional generalized tonic clonic convulsions. Her examination revealed marked hypotonia, micro-ophthalmia of right eye, microcephaly (Fig. 1), normal deep tendon reflexes and bilateral extensor plantar response. Left eye showed multiple retinal lacunae varying from ½-1 disc diameter size with clear cut margins and coloboma of the optic disc (Fig. 2). Serologic test for intrauterine infections were negative. X-ray of chest, spine and skull were normal. EEG showed intermittent paroxysmal spike and wave discharges with complete asynchrony between the cerebral hemispheres. CT scan showed abnormal separation and dilatation of lateral ventricles, sub-ependymal bulges in the medial aspect of the occipital horn of lateral ventricles, suggestive of cortical heterotopias. The third ventricle was high riding and dilated consistent with agenesis of the corpus callosum. A large low density structure in the posterior fossa typical of the Dandy Walker cyst was also seen (Fig. 3). Karytoyping showed normal 46XX pattern. At seven months of age she had received various anticonvulsants but continued to have recurrent seizures. She did not achieve any of the milestones. Blink reflex and auriculo palpebral reflex were asbent.
The present case had all the three characteristic features of Aicardi Syndrome viz agenesis of corpus callosum, chorioretinal lacunae and seizures. Callosal agenesis may be partial or complete and may be associated with other anomalies(3). The eye findings are constant and necessary for the diagnosis of Aicardi Syndrome. These findings are usually bilateral, but not all eyes are equally affected. The various ocular anomalies described include micro-ophthalmia, chorioretinal lacunae, coloboma of optic disc, persistent pupillary membrane, synechiae of iris, retinal dysplasia with total detachment, colobomatous cyst, etc.(4). The chorioretinal lacunae may vary in size from <1/10 to six times the disc diameter(4). They are well defined and may have minimal pigmentation at their borders. Histopathologically these areas consist of choroidal and retinal pigmentary epithelium atrophy with foci of papillary proliferation at the peripheries(4). This has to be differentiated from chorioretinitis of intra-uterine infection especially congenital toxoplasmosis which have poorly defined margins(5). Seizures in Aicardi Syndrome generally start early in life and have always been myoclonic in type. They may precede, accompany or followed by other seizure types as in our case. Mental retardation, hypotonia, lack of interest in visual and auditory stimuli and variable pyramidal tract signs have been observed in most of the reported cases. Numerous abnormalities including lissence-phaly, polygyria, microgyria and cortical heterotopias have been found on pathological examination of the brain(6). The most common skeletal abnormality described is fusion of the vertebral bodies. Other abnormalities include block vertebrae, hemivertebrae, butterfly vertebrae, spina-bifida occulta, scoliosis, abnormalitis in costovertebral articulations, facial and skull asymmetry and increased inter orbital distance(3). Our case did not have any of these abnormalities. Agenesis of the corpus callosum and cortical heterotopias can be seen on computed tomographic scan or magnetic resonance imaging. In addition there may be asymmetry of the cerebral hemispheres and lateral ventricles, porencephaly, arachnoid cyst, cerebellar abnormalities and cortical heterotopias and Dandy-Walker syndrome(7). Agenesis of corpus callosum, cortical heterotopias and Dandy-Walker malformation were present in our case. The presence of Dandy-Walker malformation is an unusual feature. A characteristic EEG pattern is found in all cases of Aicardi syndrome, consisting of multifocal epileptiform occurring in a burst suppression pattern, showing complete asynchrony between the two hemispheres. This EEG pattern is attributed not only to anatomical but also electrophysioloigcal disconnection between the two hemispheres and also the main subcortical nuclei which exerts a pacemake function on the cortical rhythm. Some workers have concluded that the EEG pattern is pathognomonic of Aicardi syndrome while others have questioned it(8). The exact cause of this distinct entity is unknown, X-linked dominant inheritance has been suggested(9). It is believed that this disorder is caused by a mutational event occurring during meiosis in one of the X-chromosomes. The mother is normal in all the reported cases and there is only one report of familial occurrence. This hypothesis has been supported by reports of Aicardi Syndrome in a pair of dizygotic twins. Most affected patients have 46XX karyotype. Only two cases have been reported in males. One of them had 47XXY and the other 46XY karyotype. It is likely that this disorder is lethal in hemizygous males. Some authors have speculated that the location of the gene in Aicardi syndrome is Xp 22.3 area(10). However, the exact location and nature of the gene is yet to be confirmed. Careful ophthalmological examination, electrophysiological and radiological investiga-tions are essential to differentiate this syndrome from intrauterine infection. The definitive diagnosis will help in the management of the patient, prognosticating the outcome and genetic counselling(5).
|
![]()