|
|
Case Reports Indian Pediatrics 2008;45: 595-598 |
||||||
|
Partial Trisomy 9q due to Maternal 9q/17q Translocation |
||||||
|
Sheela Nampoothiri
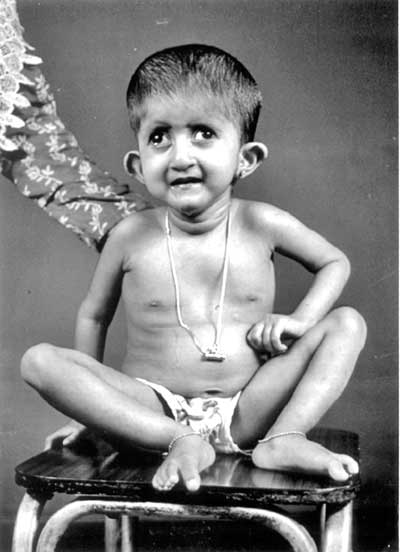
Introduction Partial trisomy 9q is a distinct phenotype with severe psychomotor retardation, dolichocephaly, long fingers and toes, prominent beaked nose, deep- set eyes and camptodactyly(1,2). We present four children with this condition who are first cousins. Case Report The proband (IV 5) was a 13 day old male baby weighing 2250 g born to a 30 year old second gravida mother by cesarean section. The parents were non consanguineous. On examination the baby had dolichocephaly, a prominent beaked nose with deep-set eyes, a small mouth, and ears with overfolded helices. The palate was narrow and high-arched. He fed poorly and had epiphora from both eyes due to associated trichiasis. He had prominent arachno-dactyly. The fingers were flexed across the thumb bilaterally. He also had flexion contractures of both knees and left elbow. The limbs were extremely thin due to reduced muscle mass (dolichostenomelia). X- rays showed thin, osteoporotic long bones. He had bilateral undescended testes. The trichiasis and flexion contractures improved gradually. At one year of age the only developmental milestones acquired were social smile and stranger anxiety. He had severe hypotonia and total head flop. CT scan showed ventriculomegaly; EEG was normal. At four years he weighed only 9 kg and was only able to crawl but had not acquired any language skills. The child died due to bronchopneumonia at the age of 4½ years. The elder sibling (IV 4) also has global developmental delay and seizures. He too has long fingers and toes and flexion contractures of knees and elbows since birth, which improved with time. He cannot flex the index fingers bilaterally. He has associated microcephaly (48 cm) and a prominent fleshy nose. The mother’s brother’s daughter (IV 6) also has a similar phenotype with long fleshy nose, long fingers and toes, microcephaly, global developmental delay, bilateral low set flared pinna with bilateral alternating divergent squint (Fig. 1). The mother’s sister (III 10) has a male child (IV 10) with the same phenotype, and additionally he has cleft palate, camptodactyly of the 2nd, 3rd, and 5th fingers on left side, contracture of both knees and right elbow joint, right inguinal hernia, right divergent squint and overfolded pinna on the left side. He had a birth weight of 2500 g, but at 3 months he weighed only 3 kg. Head circumference was 38 cm at 3 months of age.
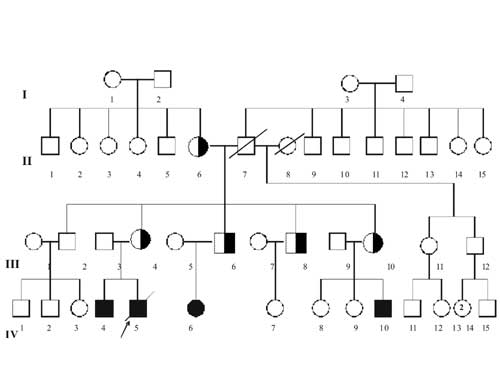
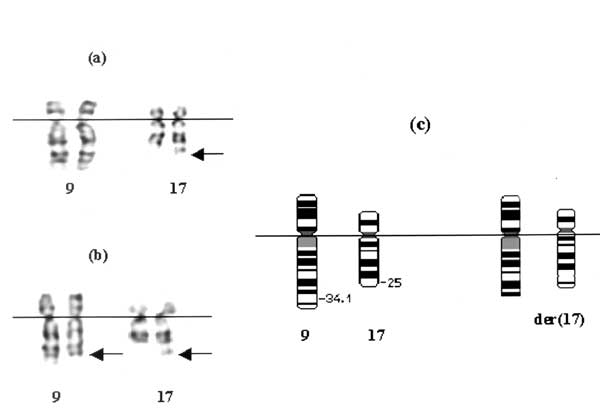
Peripheral blood samples were obtained from the proband (IV 5) and subsequently from his mother (III 4), III 6, III 8 and III 10 and II 6 (Fig. 2). Metaphase chromosomes were analyzed by standard trypsin-giemsa (GTG) banding. Chromosome analysis of the proband’s (IV 5) lymphocytes showed a derivative chromosome 17. Subsequently, a balanced trans-location was demonstrated in the mother’s lymphocytes (III 4) between chromosome 9q and 17q. The break points were 9q34 and 17q25. Thus, the maternal karyotype (III 4) was determined as 46,XX, t(9; 17) (q34; q25). Consequently, the proband’s (IV 5) chromosomal imbalance was interpreted as partial trisomy 9q34®qter and monosomy for region 17q25®qter (46,XY,der (17)t(9;17)(q34;q25) and the same unbalanced chromosomal anomaly were noted in IV 4, IV 6, IV 10. III6, III 8 and III 10 who were siblings and parents of these children with unbalanced karyotype showed a balanced translocation, 46,XX,t(9;17) (q34;q25) as III 4 (Fig. 3). Cytogenetic studies revealed that the translocation was inherited from the maternal grandmother (II 6).
Discussion Partial trisomy 9q is rare; and only 5 cases are reported till date. Chromosomal analysis of the proband and three other children with similar phenotype showed that all four of them have partial trisomy 9q and had inherited this unbalanced chromosome from their respective parents who were siblings and were carriers of a balanced t(9:17) (q34:q25) (III 4, III 6, III 10). The break point in our family was in q 34 and q25 in chromosomes 9 and 17 respectively. If the translocated segments were very small in genetic content, the resulting unbalanced gametes would lead to viable pregnancies leading to children with malformation and would not end up in miscarriages. On the contrary if the translocated segments are very big, the unbalanced gametes cannot be viable and this would lead to spontaneous abortions. In our case the translocated segments in 9q and 17 q were very small and hence it had led to birth of children with partial trisomy 9q. Even though five cases of partial trisomy 9q have been reported earlier, this is the first case where there was a balanced translocation between q arms of chromosome 9 and 17(1). The sibling (III 8) who was also found to be a carrier of this translocation. All these four siblings had inherited the translocation from their mother (II 6). This information helped III 8 to opt for prenatal diagnosis and karyotyping was performed in the amniotic fluid at 16 weeks. The fetus had a normal karyotype and they later had a normal female baby. Partial trisomy 9q usually result from unbalanced meiotic segregation of parental balanced translocation involving chromosome 9 and an autosome(1,3-5). Duplication of 9q segment due to balanced rearrangement as an inverted insertion of 9q 34.3 segment in q arm of chromosome 9 has been reported to produce the phenotype of partial trisomy 9q(6). De novo tandem duplication of 9q 22.2-q 31.1 has also shown to produce a phenotype consistent with partial trisomy 9q(7). It is highly pertinent to do a meticulous search for the trisomic segment in a patient with compatible features of partial trisomy 9q as the trisomic segment can be very small which is corresponding to the segment q 31- 32(8). The main differential diagnoses of partial trisomy 9q include congenital contractural arachnodactyly (CCA) and Marfan syndrome. In partial trisomy 9q, patients do not have the characteristic cardiac anomalies and upward dislocation of the lens, which are the hallmarks of Marfan syndrome, and they also lack the "crumpled pinna" which is typical of CCA(9,10). Marfan syndrome and CCA are autosomal dominant conditions and these conditions can be diagnosed by prenatal molecular studies. This case highlights the importance of detailed pedigree analysis while encountering patients with unbalanced chromo-somal anomalies. Contributors: SN diagnosed the condition, evaluated the family tree and has picked up other affected members and drafted the article. She will act as the guarantor of the manuscript. LRL helped in the clinical evaluation of the family. MVT performed the cytogenetic analysis for the confirmation of the diagnosis and helped in the drafting of the manuscript. AA was involved with MVT for the cytogenetic analysis. Funding: None. Competing interests: None stated. | ||||||
|
References | ||||||
|
|
![]()