The Popliteal pterigium syndrome in its lethal
autosomal recessive form, the Bartsocas Papas syndrome (BPS), was first
described in 1972(1). The chief features include a popliteal pterygium
with a cord containing nerves and vessels, synostosis of hand and foot
bones with digital hypoplasia and syndactyly, facial clefts,
ankylo-blepharon and filiform bands between the jaws(2). More than 20
cases have been reported in the literature, mostly from medi-terranean
ancestry(Greek, Italian, Spanish) but occasionally from Dutch, Arabs,
Turkish and Iranian(2-7).
We report, for the first time, patient with BPS from
a Pakistani family living in Kuwait along with review of literature.
Case Report
This boy was born to an 18-year-old Pakistani primi
mother at full-term by vaginal delivery assisted by forceps. Parents
were first-degree cousins and were healthy. There was no family history
of any congenital anomalies. The mother had one abortion at 3 months’
gestation one-year before, the cause was un-known. The antenatal period
was uneventful. Birth weight of the neonate was 2.7 kg, head
circumference 34 cm, length 47 cm and chest 32.5 cm. Apgar scores were 5
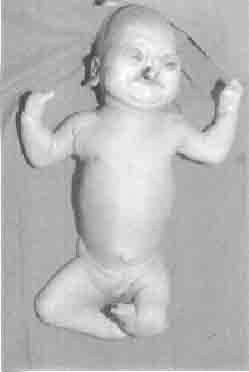
and 7 at 1 and 5 minutes respectively. The baby had strking anomalies of
face, hands, legs and genitalia (Fig. 1). There were absent hair
on scalp, eyebrows, and eye lashes with bilateral ectropion and corneal
opacities. Hypoplastic deformed nose, bilateral cleft palate and lips
and signathia (fusion of lips) were also noted (Fig. 1). Saliva
bubbles were coming out at both corners of mouth from salivary pits.
There were absent thumbs and fusion of other fingers (mitten hands) with
hypoplasia of nails. Marked shortening of both lower limbs with thick
pterygium extending from ischea to heels was noted in addition to
syndactyly of toes (all on left, and second to fourth toes on right).
The genitalia were ambiguous with small phallus and pigmented folds of
skin of scrotum. Skeletal survey showed absent first phalanx and
shortening of tibia and fibula. Ultrasound of head and abdomen were
normal. An asymptomatic soft systolic murmur was diagnosed as peripheral
pulmonary stenosis by echocardiography. Karyotype was normal. The soft
tissue fusion of lips was incised by plastic surgeon, but mouth opening
remained restricted and feeds were given by an orogastric tube. The
eyelids were stitched to prevent exposure keratitis. The baby was
discharged home at 2 months of age on gavage feeding, but suddenly died
at 4 months of age at home. The exact cause of death could not be known
as autopsy was refused.
 |
|
Fig. 1. Neonate with alopecia, facial defects,
thick popliteal pterygium, mitten hands with absent thumbs,
syndactyly of toes and hypoplastic genitalia. |
Discussion
Bartsocas and Papas reported a family, in which the
parents were third cousins, with 4 siblings (3 female, 1 male stillborn)
severely affected by BPS(1). Clinical features include popliteal
pterygium with a cord containing nerves and vessels, low birth weight,
syno-stosis of hand and foot bones with digital hypoplasia and
syndactyly; facial clefts, microcephaly, ankyloblepharon, hypoplastic
nose, filiform bands between the jaws, lower lip salivary pits and
hypoplastic genitalia(2). Our case had most of the typical features of
BPS along with the additional finding of peripheral pulmonary stenosis,
which has not been reported in literature. Various anomalies associated
with BPS include renal agenesis, esophageal atresia, hypo-plastic
diaphragm, agenesis of the shaft of the penis and anal atresia(4); and
super-numerary nipple(7); none was present in our case.
BPS should be differentiated from the lethal multiple
pterygium syndrome having generalized pterygia and which has 3 distinct
types based on bony fusion and modeling errors of bones(8); the
pterygium in BPS is localized to lower limbs.
Most cases described are among the Mediterranean
ancestry(2). Hennekam, et al. described 2 Dutch sibs(4), and
Massoud, et al. described 4 sibs in a non-consaguineous
Arab-Asian family, one was stillborn; the other 3 children lived 10 to
17 months(5). Mavilli, et al. described an 8-month-old Turkish
girl(6), and Shafeghati, et al. an Irani infant(7) with BPS. Our
case is from south-eastern-Asian ancestry (Pakistani) which has not been
described previously to our knowledge.
The exact pathogenesis of BPS is unknown. The
potential teratogenic effect of transient or persistant edema during
embryonic and fetal development was thought but absence of edema in BPS
points towards other etiologies(8). A unique and generalized vascular
problem is suggested as the disruptive process is not located in an
anatomic site(4). An alternative explanation proposed is a pleiotrophic
autosomal recessive dysplasia sequence of the "ectoderm ring" of
Blechschmidt, based on the characteristic malformations, which are
"atypical, bizarre, and ragged-at-the-edges", indicating tissue
necrosis(2). Depsite the lack of apparent consanguinity in their report
the autosomal recessive pattern can not be ruled out because of
relatively small Arab community as reported by Massoud, et al.(5).
The presence of four affected siblings and first degree consanguinity
among healthy parents in our case indicates the presence of autosomal
recessive inheritance.
Most cases die in early weeks of life, the oldest
being 8-year-old(9). Our case died at 4 months due to sudden death. The
reported causes of death are broncho-pneumonia, respiratory distress and
sepsis(2,4). Antenatal diagnosis is possible as early as first trimester
using transvaginal ultrasound(10). Due to its lethal nature, early
antenatal diagnosis and option for termination of pregnancy should be
discussed with parents, along with awareness that occasional cases may
have a good prognosis.
Contributors: RMZ was involved in patient care,
supervised this manuscript and is guarantor of the study. ALS collected
the data, reviewed the literature and prepared this manuscript. EMA
helped in reviewing the literature. RKG operated on the case and
reviewed literature.
Funding: None.
Competing interests: None stated.
