|
|
|
Indian Pediatr 2016;53:
27-31 |
 |
Classical Galactosemia
Among Indian Children: Presentation and Outcome from a Pediatric
Gastroenterology Center
|
|
Moinak Sen Sarma, Anshu Srivastava, Surender Kumar
Yachha, Ujjal Poddar and Amrita Mathias
From Department of Pediatric Gastroenterology, Sanjay
Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India.
Correspondence to: Dr Surender Kumar Yachha,
Professor and Head, Department of Pediatric Gastroenterology, Sanjay
Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226 014,
India.
Email: [email protected]
Received: January 19, 2015;
Initial review: May 14, 2015;
Accepted: October 29, 2015 .
|
Objective: To analyze the presentation and predictors of
outcome of children with galactosemia.
Methods: Analysis of clinical, laboratory,
microbiological profile and outcome of patients fulfilling the
diagnostic criteria: i) clinical setting; ii) reduced erythrocyte
Gal-1-PUT enzyme activity; and iii) unequivocal response to lactose-free
diet.
Results: 24 patients; median age of symptom onset
and diagnosis: 10 (3-75) d and 55 (15-455) days, respectively. 71% had
uncorrectable coagulopathy; 71% systemic infections; and 54% had ascites.
Outcome: consisted of 87.5% survival with normalization of liver
function tests at 5.5 (1-24) months follow-up.
Conclusion: Despite delayed referral, high
Pediatric end-stage liver disease scores and systemic infections,
long-term outcome in galactosemia is rewarding. A subset of children
have developmental delay.
Keywords:Galactose, lactose-free diet, Outcome.
|
|
C
lassical galactosemia is an autosomal
recessive disorder of galactose metabolism occurring due to deficiency
of the enzyme galactose-l-phosphate uridyl transferase (Gal-1-PUT), and
responding to a galactose restricted diet [1]. In the absence of this
enzyme, galactose is converted into toxic by-products (galactitol,
galactose-1-phosphate and galactonate) that affect the liver, brain,
kidneys, lens and gonads. There is scarcity of data on clinical profile
and natural history among Indian children, resulting in lack of
awareness of this potentially treatable condition. We studied the
presentation and predictors of outcome of children diagnosed to have
galactosemia.
Methods
We analyzed the data of children with
confirmed galactosemia from July, 2003 to June, 2014 admitted in
the Pediatric Gastroenterology department of our Institution, a large
referral hospital in Northern India. Enrolled patients fulfilled all
three diagnostic criteria: (i) clinical features suggestive of
galactosemia, (ii) reduced or undetectable erythrocyte Gal-1-PUT
enzyme activity, and (iii) unequivocal response to lactose-free
diet. We retrieved the clinical, laboratory features and
follow-up data from hospital electronic records. We traced majority of
our patients telephonically for a fresh visit to document the current
status at the time of analysis. At admission, all children underwent
routine blood tests and screening for sepsis. Neutrophilia and
leucocytosis were interpreted as per the age-related cut-offs (maximum
limit of range) [2].
Diagnostic paracentesis was done in all patients with ascites. Presence
of cataract was confirmed by the ophthalmologist with direct
ophthalmoscopy. While on lactose containing diet at admission, three
samples of urine were tested for presence of non-glucose reducing
substances by Benedict’s test (glycosuria ruled out by urine dipstick
method). Gal-1-PUT assay was done by spectrofluorometry (quantitative
Beutler test) [3]. Normal values of Gal-1-PUT varied between 20-50 U/gHb.
Levels £10 U/gHb
are considered confirmatory of galactosemia. Values <5 U/gHb (lowest
laboratory limit) were reported undetectable. The test was reconfirmed
after 12 weeks of initial presentation in children who had earlier
received packed red cell transfusion or had presented with hemolysis.
Percutaneous liver biopsy was done at admission in all patients in whom
the coagulopathy corrected and ascites resolved. Additionally, upper
gastrointestinal endoscopy was performed as indicated. Pediatric
end-stage liver disease (PELD) scores were calculated as per standard
formula and scores of 17 and 25 were taken as cut-offs for comparison of
various parameters [4,5]. Systemic infection was defined as any one or
more of the following: (a) bacterial or fungal culture positivity
of blood and/or urine, (b) pneumonia on chest X-ray, (c)
cerebrospinal fluid analysis suggestive of pyogenic meningitis, (d)
spontaneous bacterial peritonitis (SBP) or culture negative neutrocytic
ascites (CNNA). SBP was defined as absolute neutrophil count
³250 cells/mm3
and ascitic fluid culture positivity. CNNA was defined as absolute
neutrophil count ³250
cells/mm3 with sterile
ascitic fluid culture [6]. Presumed infection in a sick child was
defined as high clinical suspicion with neutrophilic leukocytosis with
(out) thrombocytopenia (platelet count <100,000/mm3)
with (out) positive semi-quantitative C-reactive protein (>6 mg/dL) but
sterile body fluid cultures.
All patients were counseled and discharged on
lactose-free diet and supplements. Patients were thereafter periodically
followed up. Patients with at least 6 months of follow-up were analyzed
for outcome. Compliance to lactose-free diet, clinical improvement, and
time taken for normalization of liver function tests were assessed.
Normal liver function tests was defined as normal albumin, international
normalized ratio (INR) and transaminases <2 times upper limit of normal.
Surgery was advised by the ophthalmologist if the cataract status was
dense, persisted despite diet-compliance or if the child was at risk of
amblyopia at 1-3 months of follow-up. Children older than 18 months of
age were subjectively assessed for development in all domains.
Statistical analysis: For comparison between two
groups, we used chi-square test for categorical variables. The clinical
and laboratory factors associated with outcome were analyzed by a
logistic regression analysis. SPSS version 16.0 (SPSS Inc, Chicago, IL,
USA) was used for all statistical analysis, and a P value of
<0.05 was taken as significant.
Results
Out of 1189 neonatal cholestasis patients, 24
children (16 boys) were diagnosed to have galactosemia. Overall median
age of onset of symptoms and age at diagnosis (age at dietary
intervention) was 10 (3-75) d and 55 (15-455) d, respectively. There was
a median delay in diagnosis of 45 (12-380) d. All had history of
neonatal jaundice. Majority had uncorrectable coagulopathy and ascites (Table
I).
TABLE I Clinical Profile of Patients with Galactosemia at Admission (N=24)
|
Clinical profile |
n (%) |
|
Symptoms$ |
|
|
Poor feeding |
16 (67) |
|
Lethargy |
15 (53) |
|
Generalized tonic clonic seizures |
7 (29) |
|
Signs |
|
|
Splenomegaly |
24(100) |
|
Ascites |
13 (54) |
|
Bilateral cataract |
13 (54) |
|
Other features |
|
|
Uncorrectable coagulopathy* |
17 (71) |
|
Recurrent hypoglycemia |
15 (63) |
|
Transient hemolysis# |
4 (16) |
|
Sibling deaths |
9 (38) |
|
Consanguinity |
3 (13) |
|
$Jaundice and hepatomegaly present in all children,
*International normalized ratio (after vitamin K injection) >
1.5, #Hemoglobin level below age specific cut-off [2],
reticulocyte count >2% and peripheral smear suggestive of
hemolysis. |
Liver function tests profile (median with range)
showed total/direct bilirubin: 10.8 (2.8-24)/5.0(1.6-12.0) mg/dL,
aspartate/ alanine aminotransferase: 191 (52-861)/84(26-525) U/L; serum
albumin: 2.7 (1.9-4.2) g/dL; alkaline phosphatase: 937(143-1464) U/L;
gamma-glutamyl transpeptidase: 24 (8-818) (U/L), and international
normalized ratio (after vitamin K): 1.7 (1.0-6.8). Positive urinary
non-glucose reducing substance samples ( ³2
of 3) were seen in 22 cases. Only two children had Gal-1-PUT levels of
8.7 and 10 U/gHb; rest 22 had undetectable levels. Twelve of 14 liver
biopsies done showed cirrhosis or bridging fibrosis; 2 had
macrovesicular steatosis. Median (range) PELD score at diagnosis was 24
(9-51).
TABLE II Profile of Infections First at Admission And Readmission in Non-compliant Patients
|
Type of infection* |
n (%) |
Details |
|
Systemic infection# |
17 (71) |
|
|
Blood culture positive* |
9 (38) |
E.coli (n=3), CONS (n=2), Klebsiella (n=1), Pseudomonas (n=1),
Citrobacter (n=1), Methicillin-resistant S. aureas (n=1) |
|
Urine culture positive* |
7 (29) |
E.coli (n=4), Candida (n=3) |
|
Respiratory infection |
1 (4) |
Lobar pneumonia on chest X-Ray (left upper lobe) |
|
Pyogenic meningitis |
1 (4) |
CSF culture negative |
|
SBP or CNNA |
3 (12.5) |
E.coli (n=1), ascitic fluid culture negative (n=2) |
|
Presumed infection |
4 (17) |
Not applicable |
|
#Multiple site infections :Blood and urine
culture positive (3); blood culture positive and CNNA (1); CNNA
and meningitis (1); blood culture positive and lobar pneumonia
(1). *4 readmitted non-compliant patients had Pseudomonas
and Citrobacter in blood culture (1 each) and E.coli in urine
culture (2); CNNA: Culture-negative neutrocytic ascites; SBP:
spontaneous bacterial peritonitis. |
Table II shows the composite infectious
profile of all infected patients. 13 children with systemic infection
had a definitive infective focus at admission. Additionally 4 infants
initially stable were readmitted with systemic infection at follow-up as
they were non-compliant to LFD. 11/17 (68%) systemic infection had a
gram negative infection, mostly with Escherichia coli (n=8).
Symptom onset <2 weeks of age (n=5) was significantly
associated with systemic infection (P=0.002) than those with >2
weeks (n=8). PELD ³25
at first admission was significantly associated with systemic infection
(p=0.04, or 7.0, 95% CI 1.04-46.9).
Fig. 1 shows the natural history of
galactosemia patients on follow-up. Mean days of hospitalization was 12
(4-23) d. Long term follow-up ( ³6
months) was available in 18 patients who had good LFD compliance and
were analyzed for outcome. LFT improved in 4.5 (1-18) and 15 (4-24) mo
in those diagnosed <4 weeks (n=12) and >4 weeks (n=6) of
age, respectively (P=0.02). No difference in LFT normalization
was seen with PELD cut-off scores of 17 or 25.
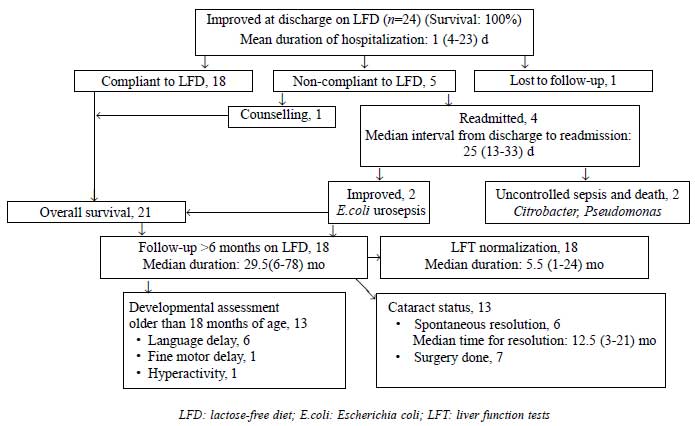 |
|
Fig. 1 Clinical course and outcome of
galactosemia patients.
|
One child operated at 8 months was permanently blind
due to irreversible amblyopia. Thirteen children older than 18 months
age were assessed for development. Of 6 with initial language delay, two
showed catch-up at subsequent follow-up. Hypodensities in white matter
on imaging with persistent fine motor delay at 24 months was noted in
one child.
Discussion
This composite study reported the presentation,
natural history and predictors of outcome in galactosemia among Indian
children. Though the exact Indian prevalence is not known, galactosemia
constituted 2% of all our neonatal cholestasis referrals. In an analysis
of 1008 cases of neonatal cholestasis, galactosemia constituted 4% of
all cases and 35% in the metabolic liver disease sub-group [7]. Our
median age of diagnosis was comparable to that reported by Singh, et
al. [8]. Delay in diagnosis resulted in higher hepatocellular
dysfunction (100% vs. 64-89%), systemic infection (71% vs.
13-40%), cataract (54% vs. 13-30%) and seizures (29% vs.
3-17%) in our study compared to other series where early referral was
attributable to neonatal screening [9,10]. In the study by Honeyman,
et al. [10], 95% neonates were started on LFD by day 30 of life .
E.coli culture positivity was higher in our
series (47% vs. 24%) compared to Waggoner, et al. [9].
Transplecental deficiency of IgM bactericidal opsonic
antibodies-complement system in neonates and inhibition of leucocyte
bactericidal activity by accumulated galactose predispose to
gram-negative infections [11,12].
The disease has a favorable prognosis by timely
referral, introduction of LFD, and long term compliance; Reintroduction
of lactose in such cases may be fatal [13]. LFT improved by median
interval of 5.5(1-24) months of starting therapy, significantly earlier
in those diagnosed <4 weeks. PELD cut-off’s >17 (United Network for
Organ Sharing Status for liver transplant) and >25 (high incidence of
death rate: 4.6/1000 patient-years) did not influence the improvement in
LFT at follow-up [3,4]. Spontaneous resolution of cataract was not
related to age at diagnosis or dietary intervention. This is in contrast
to Waggoner, et al. [9] who showed that 90% cataracts resolve
spontaneously if dietary intervention is begun at mean of 77 days of age
[9]. Given the risk of irreversible ambylopia, early cataract surgery
(1-8 weeks of life) is presently recommended by most pediatric
ophthalmologists as performed in seven of our cases who continued to
have dense cataract despite four weeks of LFD [14].
Bosch, et al. [15] found subnormal cognitive
outcomes in galactosemia children older than six years age, despite
strict adherence to diet [15]. Small number of patients and
retrospective design were the main limitations of our study. As a
result, we could not assess IQ, use development scoring systems
or identify risk factors for delayed milestones.
A cholestatic neonate with ascites, coagulopathy,
seizures, family history of sibling death, consanguinity and/or
hemolysis should raise a suspicion for galactosemia. Despite high PELD
scores (advanced disease) and systemic infections, this condition is
salvageable with lactose-free diet. Neonatal screening or early
diagnosis are helpful strategies to having a favorable outcome.
Contributors: MSS: data acquisition,
interpretation and analysis; drafting of manuscript. AS and SKY:
Critical revision for intellectual content. UP: Study supervision. AM:
Technical expertise. All authors approved the final version.
Funding: None; Competing interest: None
stated.
|
What This Study Adds?
• There is a considerable delay in referral
of galactosemia patients in India.
• Good clinical outcome is seen despite high PELD scores and
systemic infections at presentation.
|
References
1. Applegarth DA, Toone JR, Lowry RB. Incidence of
inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics.
2000,105:e10.
2. Gajjar R, Jalazo E. Hematology section. In:
Engorn B, Flerlage J, editors. The Harriet Lane Handbook. 20th ed.
Philadephia: Elsevier-Saunders; 2014. p.345.
3. Fujimoto A, Okano Y, Miyagi T, Isshiki G, Oura T.
Quantitative Beutler test for newborn screening of galactose using
fluorometric microplate reader. Clin Chem. 2000;46:806-10.
4. Barshes NR, Lee TC, Udell IW, O’Mahoney CA, Karpen
SJ, Carter BA, et al. The pediatric end-stage liver disease
(PELD) model as a predictor of survival benefit and post-transplant
survival in pediatric liver transplant recipients. Liver Transpl. 2006;12:475-80.
5. McDiarmid SV, Merion RM, Dykstra DM, Harper AM.
Selection of pediatric candidates under the PELD system. Liver Transpl.
2004;10:S23-30.
6. Runyon BA. Ascites and Spontaneous Bacterial
Peritonitis. In: Feldman M, Friedman LS, Brandt LJ, editors.
Sleisenger and Fordtran Gastrointestinal and Liver Disease.
Pathophysiology/ Diagnosis/ Management. 9th ed. Philadephia: Saunders
Elsevier; 2010.p.1517-42.
7. Consensus Report on Neonatal Cholestasis Syndrome.
Pediatric Gastroenterology Subspecialty Chapter of Indian Academy of
Pediatrics. Indian Pediatr. 2000;37:845-51
8. Singh R, Thapa BR, Kaur G, Prasad R. Biochemical
and molecular characterization of GALT gene from Indian galactosemia
patients: Identification of 10 novel mutations and their structural and
functional implications. Clinica Chimica Acta. 2012;414:191-6.
9. Waggoner DD, Buist NR, Donnell GN. Long-term
prognosis in galactosaemia: results of a survey of 350 cases. J Inherit
Metab Dis. 1990;13:802-18.
10. Honeyman MM, Green A, Holton JB, Leonard JV.
Galactosaemia: results of the British Paediatric Surveillance Unit
Study, 1988-90. Arch Dis Child. 1993;69:339-41.
11. Stoll BJ. Infections of the Neonatal Infant.
In: Kliegman RM, Stanton BF, Schor NF, St.Geme III JW, Behrman RE,
editors. Nelson Textbook of Pediatrics. 19th ed. Philadephia: Elsevier-
Saunders; 2011.p. 629-48.
12. Kumar M, Yachha SK, Gupta RK. Neonatal
cholestasis syndrome due to galactosemia. Ind J Gastroenterol.
1996;15:26-67.
13. Walter JH, Collins JE, Leonard JV.
Recommendations for the Management of Galactosaemia. UK Galactosaemia
Steering Group. Arch Dis Child. 1999;80:93.
14. Wright K, Lens abnormalities. In: Wright
K, Spiegel PH, editors. Pediatric Ophthalmology and Strabismus. 2nd ed.
New York: Springer-Verlag; 2003.p. 450-80.
15. Bosch AM, Grootenhuis MA, Bakker HD, Heijmans HS,
Wijburg FA, Last BF. Living with classical galactosemia: health-related
quality of life consequences. Pediatrics. 2004;113:e423-8.
|
|
|
 |
|
