|
|
|
Indian Pediatr 2012;49: 58-59
|
 |
Microvillous Inclusion Disease Diagnosed by Gastric Biopsy |
|
Niranjan Thomas, *Anna Benjamin Pulimood, Manish Kumar and Atanu Kumar Jana
From the Department of Neonatology and * Department of Gastrointestinal Sciences, Christian Medical College,
Vellore 632004, Tamil Nadu, India.
Correspondence to:
Dr Niranjan Thomas,
Department of Neonatology,
Christian Medical College Hospital,
Vellore 632004, Tamilnadu, India.
Email:
[email protected]
Received: June 22, 2010;
Initial Review: August 25, 2010;
Accepted: September 13, 2010.
|
Protracted diarrhea in neonates is uncommon and usually requires an intestinal biopsy for etiological diagnosis. Gastric biopsy has not been used in the routine diagnosis of this condition. We report the first documented patient with microvillous inclusion disease from India, where the diagnosis was established by a gastric biopsy.
Key words: Diagnosis, Gastric biopsy, Infant, Microvillous inclusion disease.
|
|
Neonatal onset of protracted diarrhea is rare and
the differential diagnosis includes congenital
carbohydrate malabsorption disorders,
congenital ion transport defects, infectious or post infectious enteropathies, autoimmune or allergic enteropathies, IPEX syndrome, microvillus inclusion disease, and tufting enteropathy [1].
Microvillus inclusion disease (MVID) is an autosomal recessive disorder that presents in the neonatal period with severe secretory diarrhea and has no specific treatment and a high mortality [2]. The diagnosis of this condition is based on typical light and electron microscopic (EM) changes seen on small intestinal biopsies. We report the first child with MVID from India where the diagnosis was based on an antemortem gastric biopsy, confirmed later by a post mortem intestinal pathological examination.
Case Report
This female baby was the third born child to healthy non consanguineous parents. The previous two babies were born by caesarean section (LSCS) at term gestation, developed profuse watery diarrhea on day three of life, and died on day three and eight of life, respectively with no specific diagnosis. The mother had an uneventful antenatal period and ultrasound done at 35 weeks gestation showed dilated bowel loops and increased amniotic fluid volume. She developed preterm prelabor rupture of membrane at 35 weeks and delivery was by LSCS in view of the previous two LSCS. This near term appropriate for gestational age girl baby, weighing 2320 g was admitted to nursery for observation in view of two previous early neonatal deaths. She was well for the first two days of life but developed profuse watery diarrhea with signs of severe dehydration on day 3. She was kept nil per oral, given volume expansion with normal saline, started on fluid replacement therapy and antibiotics after a sepsis workup. Investigation showed normal blood sugar and serum electrolytes with severe metabolic acidosis. Stool reducing substance was negative. Work up for inborn errors of metabolism and sepsis was negative. After correction of dehydration and metabolic acidosis, she was started on lactose and sucrose free diet but continued to have watery diarrhea. She was kept nil oral and given total parental nutrition and investigated for a secretory diarrhea as her stool output continued to be high despite not being fed. The possibility of congenital ion transport defect was ruled out based on the stool sodium (35 mmol/L) and chloride content (21 mmol/L).
Considering a possibility of microvillus inclusion disease, a duodenal biopsy was planned and an endoscopy was performed. However, the endoscope could not be passed beyond the pylorus and only a gastric biopsy was done. On light microscopy, the gastric mucosa showed severe glandular atrophy. The surface and foveolar epithelium was short columnar and showed marked mucin depletion, focal intercellular edema, mild megalocytic change and focal marked cytoplasmic pallor in some cells and vacuolation in others.Ultrastructural study of the gastric mucosa showed focal absence of microvilli on the surface or foveolar epithelial cells with few to many cytoplasmic vacuoles containing well-formed or degenerate microvilli confirming a diagnosis of microvillus inclusion disease. In some cells the central part of the luminal surface with its microvilli appeared invaginated while the lateral edges protruded into the lumen along with the edges of adjacent cells. Vacuoles containing microvilli were seen in the cytoplasm underlying the invaginated area. In other cells a thin cytoplasmic flap was seen to cover vacuoles close to the surface with microvilli only on the abluminal side (Fig. 1). The vacuoles with less preserved microvilli and those of a similar size but containing structure-less material were located deeper in the cytoplasm than those with well-formed microvilli.
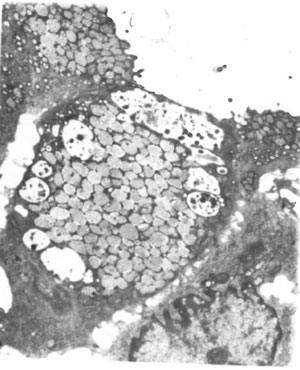 |
Fig. 1 Electron micrograph of a gastric surface epithelial cell with a cytoplasmic flap on the luminal side enclosing a vacuole with microvilli on the abluminal surface, and other cytoplasmic vacuoles containing microvilli and structureless material (Uranyl acetate and lead citrate; Final magnification x 2233). |
In view of the poor long term prognosis and lack of facilities for a small bowel transplant, the parents opted to withdraw support. Baby died on day twenty three of life. An autopsy was performed and the intestinal tissue studied and stained with hematoxylin and eosin, Periodic acid-Schiff (PAS), CD 10 and carcinoembryonic antigen (CEA). The surface epithelium was preserved in small foci and showed absence of villi and the PAS stain as well as the immunohistochemical stains for CD 10 and CEA revealed absence of an observable brush border with increased staining at the apex of the enterocyte consistent with microvillus inclusion disease.
Discussion
Protracted diarrhea in the neonatal period is an uncommon clinical problem with a variety of underlying etiologies. Our patient presented with watery diarrhea with severe dehydration in the early neonatal period and had a family history of two siblings dying of similar symptoms in the first week of life. Carbohydrate malabsorption was ruled out as stool reducing substances were negative and the infant continued to have loose stools while on a carbohydrate free formula. The clinical picture of continuous diarrhea despite not being fed is suggestive of a secretory diarrhea.
The causes of secretory diarrhea in the neonatal period can be categorized into those with normal villous architecture and those with villous atrophy, based on intestinal biopsy [1]. Congenial ion transport disorders like sodium-hydrogen exchanger and congenital chloride/sodium diarrhea belong to the former group. These were ruled out in our patient based on the stool and serum electrolyte concentrations. Those with villous atrophy are differentiated based on the immuno-histopathology and electron microscopy findings on the intestinal biopsy.
These findings have been well described in the small and large bowel, but there are few reports in literature on their presence in the gastric mucosa [3]. In our patient, we could establish the diagnosis of MVID by electron microscopy of the gastric mucosa suggesting an alternative to duodenal biopsies that may be technically difficult in neonates. The ultrastructural findings in the gastric mucosa of our patient were very focal, suggesting that a careful and extensive search for microvillous inclusions is essential. The range of changes seen parallels that described in intestinal biopsies, and our findings confirm that these inclusions are probably formed by invagination of the luminal surface and closure of invaginated foci with cytoplasmic flaps rather than by arrested transport towards the luminal surface [4].
The presence of antenatal dilated bowel with increased amniotic fluid volume seen in our patient is commonly seen in ion transport disorders like congenital chloride diarrhea but has also been described in isolated cases of MVID [5]. Other than supportive measures, various drugs like loperamide, somatostatin, corticosteroids and cholestyramine have been tried with no benefit. Small bowel transplantation or small bowel and liver transplantation have been successfully used recently in this condition [6].
We report the first case with MVID from India and highlight the fact that gastric mucosal examination can be used in its diagnosis if a duodenal biopsy is not possible. Any child with a neonatal onset severe diarrhea should undergo early intestinal biopsy after ruling out carbohydrate malabsorption and ion transport disorders by non invasive tests.
Acknowledgments: Dr Pierre Russo, Chief of Anatomic Pathology at The Children’s Hospital of Philadelphia, USA, for his expert opinion and confirming the diagnosis.
Contributors: NT, MK and AKJ were involved in the diagnosis and management of the case. ABP gave the final pathological confirmation. NT prepared the manuscript and will act as guarantor.
Funding: None; Competing interests: None stated.
References
1. Cutz E, Rhoads JM, Drumm B, Sherman PM, Durie PR, Forstner GG. Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med. 1989;320:646-51.
2. Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr. 2004;38:16-26.
3. Schofield DE, Agostini RM Jr, Yunis EJ. Gastrointestinal microvillus inclusion disease. Am J Clin Pathol. 1992;98:119-24.
4. Reinshagen K, Naim HY, Zimmer KP. Autophagocytosis of the apical membrane in microvillous inclusion disease. Gut. 2002;51:514-21.
5. Kennea N, Norbury R, Anderson G, Tekay A. Congenital microvillus inclusion disease presenting as antenatal bowel obstruction. Ultrasound Obstet Gynecol. 2001;17:172-4.
6. Ruemmele FM, Jan D, Lacaille F, Cézard JP, Canioni D, Phillips AD, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation. 2004;77:1024-8.
|
|
|
 |
|
