|
|
Case Reports Indian Pediatrics 2006; 43:61-64 |
||||
|
Carmi Syndrome Complicated by Pharyngo-Esophageal Perforation |
||||
|
Abstract: Key words: Carmi Syndrome, Congenital pyloric atresia, Epidermolysis bullosa, Pharyngo-esophageal perforation.
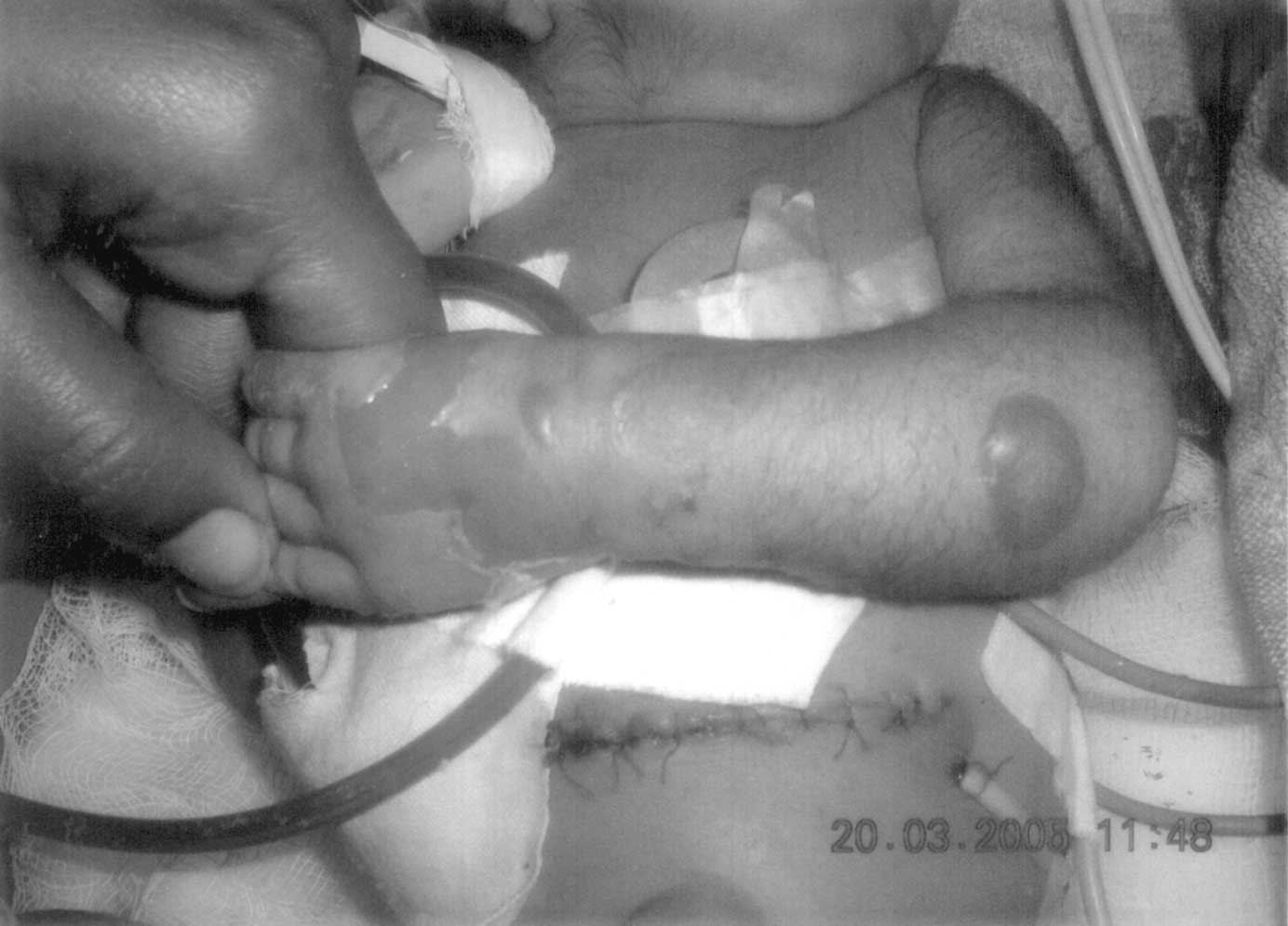
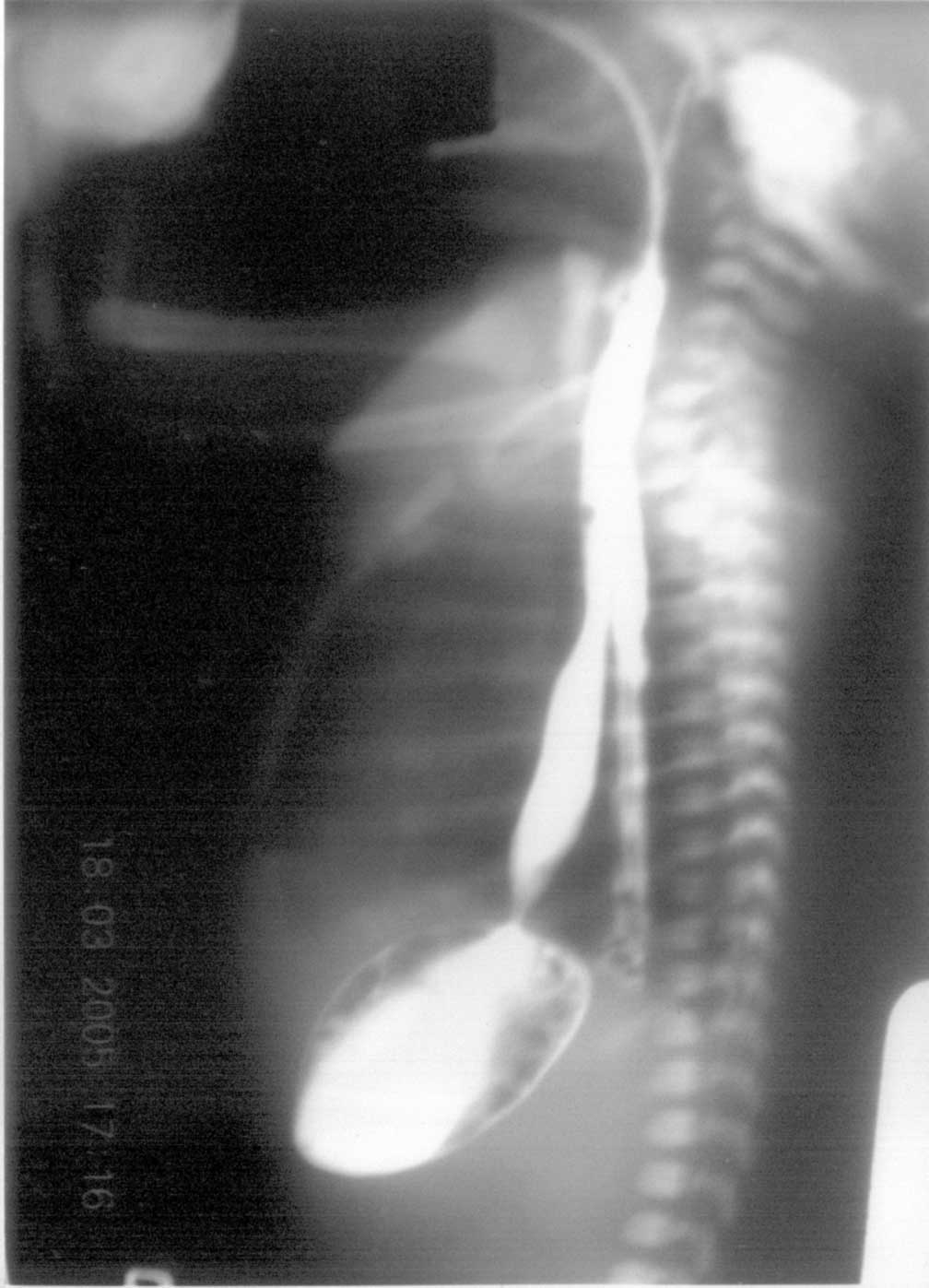
The association of CPA and EB (Carmi syndrome) was first described by Swinburne and Kolher in 1968(1). The incidences of CPA and EB have been quoted as 1 of 100,000 live births and 1 in 300,000 live births respectively(2,3); the coexistence of CPA and EB is extremely rare. Carmi Syndrome has had a universally fatal outcome previously with only few isolated survivors(3). We report an index case. Case report A 5-day-old, pre-term female neonate weighing 2.210 kg, born to G6P4 mother through normal vaginal delivery at home at 32-week gestation, was referred to us with complaints of non-bilious vomiting since birth. The parents were first cousins and had three normal siblings before her. Detailed family history revealed history of skin lesions and infantile deaths. Three siblings of patient’s mother (out of twelve) and four siblings of patient’s maternal grandmother (out of eighteen) had skin lesions, which developed just after birth and all these infants died in first three months. On examination, she had multiple bullous skin lesions over the lower extremities suggestive of EB. There was bloodstained froth emanating from the mouth, although no mucosal lesions were seen in the mouth. Upper abdomen was mildly distended. Multiple attempts to pass naso-gastric tube failed. Babygram showed coiled feeding tube in chest with a distended gastric bubble; the rest of abdomen was gasless. The upper GI-contrast study that was done elsewhere was suggestive of PEP and CPA (Fig.1). The patient underwent an emergency surgery after initial stabilization. At laparotomy, type II CPA was confirmed. Rest of the gut and other viscera were normal. Excision of atretic pylorus and gastro-duodenostomy was performed. A naso-jejunal tube could be passed across the anastomosis for early feeding. Gastric decompression was achieved by gastrostomy.
The post-operative period was stormy. The patient developed multiple new lesions over trunk and extremities over the next two days (Fig. 2). On 3rd postoperative day, patient developed features of overwhelming sepsis and respiratory distress syndrome. Despite a change in antibiotics, transfusion of blood components, ventilatory support, care of skin lesions and supportive care, her condition deteriorated and she died on 5th day.
Discussion CPA constitutes only 1% of all reported gastrointestinal atresias. Three distinct forms exist-as an isolated case, in association with other GI atresias, or in associations with EB and/or aplasia cutis congenita(4,5). EB is a rare autosomal recessive genodermatosis characterized by separation between basal cell junction and basement membrane. It is classified into three major categories: junctional EB (JEB), EB simplex (EBS), and dystrophic EB. All three types have been described with CPA. Previously, families have been known to have EB or EB-CPA complex over generations(8), but the present case is peculiar for the fact that although the previous generations had only subjects having EB without co-existing CPA, this neonate in the third generation had full-blown fatal EB-CPA complex. This probably suggests the separation of epithelium from its basement membrane as primary event manifesting in developing pylorus as CPA and strengthens the hypothesis proposed by Chang that CPA occurs as a result of an intrauterine complication of EB where the pyloric mucus membrane is affected, leading to sloughing with subsequent scarring and fibrosis and obliteration of the pyloric canal(9). The clinical course of our patient was complicated by PEP. PEP has been reported to be iatrogenic in most of the cases previously; the exact cause of this complication could not be made out here. When there is either submucosal tear, or the leak is confined to mediastnum, the condition clinically mimics EA. Differentiating the two conditions is important, as the management of these perforations is largely conservative, whereas EA will require operative correction. There are a few subtle differences in presentation, which may clinch the diagnosis. Unlike EA, there will not be any history of maternal polyhydramnios. Late presentations, blood stained secretions from oral cavity are common with pharyngo-esophageal perforations. In our patient, the blood stained discharge could have had its origin both from the mucosal lesions of EB and PEP. Radiological signs (pneumothorax, pyo-pneumothorax, retropharyngeal or sub-cutaneous air) and the position and course of nasogastric tube may help in diagnosis. It may get coiled in neck due to cricopharyngeal spasm or may course through perforation and get arrested at diaphragm to one or other side or it may pass into stomach bypassing perforation(10). A water-soluble contrast study, which is not routinely advocated in patients with CPA or EA, may be indicated when we suspect PEP. This study may demonstrate pharyngeal pseudo-diverticulum, leak in posterior mediastinum parallel to the opacified esophageal lumen (as seen in our case) or spillage of dye into pleural cavity due to free perforation. The management of PEP is conservative. If a nasogastric tube can be negotiated into stomach (either under fluoroscopic guidance), nasogastric feedings can be started. Failure of this procedure warrants parenteral nutrition or feeding gastrostomy. An oral contrast study is performed after 7-10 days and oral feedings are started once healing of esophagus is demonstrated. Surgical options available for CPA are web excisions with or without pyloroplasty, atresia excision and gastro-duodenostomy. Stamm gastrostomy can be supplemented. Recently, pyloric sphincter reconstruction by gastric and duodenal mucosa cul-de-sacs advancement and end-to-end anastomosis has been described for type II CPA(11). Many newborns with CPA succumb to sepsis or dehydration and electrolyte imbalance. Those infants who survive need close monitoring for the development of obstructive uropathy, failure to thrive, protein-losing enteropathy, respiratory compromise, and increased susceptibility to invasive infections(12). The prognosis of an isolated PEP, however, is excellent. The purpose of the manuscript is to aware the pediatricians of CPA, an entity that has familial transmission and recurrence risk in future pregnancies close to 25%. Contributions: YKS managed the case, finalized the draft and would stand as guarantor. NGN reviewed the literature and prepared the manuscript. Competing interests: None. Funding: None.
| ||||
|
References | ||||
|
![]()