|
Case Reports |
Indian Pediatrics 2000;37: 85-88 |
Familial Neuroblastoma |
| Kusumakumary P., Ravindran Ankathil,
Priyakumari T., Krishnan Nair From the Regional
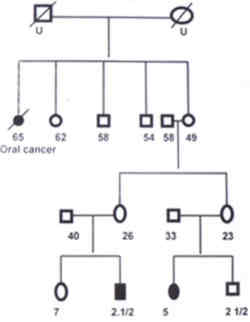
Cancer Center, Trivandrum 695 001, India. Neuroblastoma is the common extracranial solid tumor in children. The cause of neuro-blastoma is unknown in most cases. No prenatal or postnatal exposure to drugs, chemicals or radiation has been associated unequivocally with an increased incidence of neuroblastoma. Neuroblastomas usually occur sporadically, but familial incidences have been reported(1,2). This indicates that some patients exhibit a genetic predisposition to develop neuroblasto-mas. We have come across two families-in one family two siblings were affected and in the other first cousins were affected by neuro-blastoma. The clinical features of these four patients and a discussion on genetic aspects of neuroblastomas are included in this report. Case Reports Family 1 Patient I: A 9-month-old female patient was brought to us in November 1992 with one month history of irregular fever, abdominal distension and proptosis of left eye. On examination she was found to have proptosis and periorbital ecchymosis of left eye, swelling of left parietal bone and a hard nontender fixed nodular mass in the left hypochondirum crossing the midline. CT scan confirmed a mass in the paravertebral region below the left kidney, extending downwards to left iliac fossa. Laparotomy showed a large unresectable left paravertebral tumor which was biopsied and the histopathological report was neuroblastoma. Bone scan revealed increased uptake over left parietal bone and marrow aspirate was positive for neuroblastoma cells. The patient was categorized as having stage IV disease. Treatment consisted of 6 cycles of chemo-therapy with injection vincristine, cyclophos-phamide and doxorubicin at 3 weekly intervals. Tumor was found to be unresectable after 6 courses of chemotherapy. Parents shifted their residence to some other state and were lost for follow up. The child was brought along with her sibling after 5 years and re-evaluation did not reveal any evidence of disease. Patient II: In June 1997, 3-year-old younger brother of patient I was brought with complaints of progressive pallor, irregular fever, pain right leg and ecchymosis around both eyes. On general examination he was found to have anemia, periorbital ecchymosis, scalp swelling over left temporal region and bilateral cervical lymphadenopathy. Abdominal examination revealed a mass in right lumbar region which was hard in consistency. CT scan showed mixed density masses in both suprarenal region with areas of calcification. Biopsy from the scalp swelling was diagnostic of neuroblastoma. Skeletal survey revealed lytic lesions of left femur and sutural diastasis in skull bones. Bone marrow examination showed infiltration with neuroblastoma cells. Parents refused chemotherapy and the patient was lost for follow up. The pedigree of these 2 patients is shown in Fig. 1.
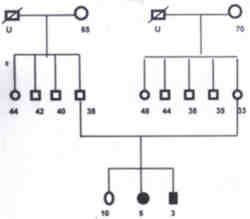
Fig. 1. Pedigree of Neuroblastoma family 1. Family 2 Patient III: A 5-year-old girl was brought on 7-1-98 with the complaints of multiple scalp swelling of one month duration and swelling of right side of neck of 2 weeks duration. On general examination she was found to have pallor, multiple scalp swelling and right cervical adenopathy. Abdominal examination revealed moderate hepatomegaly and a mass in the right hypochondrium. Ultrasound and CT scan abdomen showed a fairly large mass of mixed echogenicity in the right suprarenal area, multiple paraaortic nodes and multiple metastasis in the liver. Skeletal survey and bone scan showed evidence of disease in the skull bones. Biopsy from the node was consistent with neuroblastoma. Bone marrow was infiltrated with neuroblastoma cells. Parents refused treatment and the child was lost for follow up. Patient IV: A 2�-year-old male child (1st cousin of patient No. III) was brought on 12-2-98 with pain right leg of 2 weeks duration and swelling of left temporal region and right cervical region of 1� week duration. He had mild pallor. Lymph nodes were palpable 2�2 cm on right side of neck. Bony swelling 5�5 cm was present over left temporal region. Systemic examination did not reveal any abnormality except mild hepatomegaly. Biopsy from the lymph node was consistent with neuroblastoma. Skeletal survey showed sutural separation and lytic lesions of skull bones. Ultrasound abdomen did not reveal any abnormality. CT scan abdomen showed a 2.5 cm�3 cm partly hypoechoic mass in left suprarenal area. Bone marrow examination showed evidence of disease. Patient was on combination chemotherapy with injection cisplatin, adriamycin, endoxan and VP-16. Lymphnodes and skull swellings regressed after 2 months of chemotherapy. Child was taken abroad as the parents were working there. The pedigree of this family is shown in Fig. 2.
Fig. 2. Pedigree of Neuroblastoma family 2. Discussion Instances of familial neuroblastoma are being reported with increasing frequency. Kushner et al.(2) in their review of literature reported 23 familial aggregations of neuro-blastoma and or ganglioneuroblastoma. Relationship of affected subjects included siblings, monozygotic twins, half siblings, cousins and parent child. Draper et al.(3) had reported three families in which both children were diagnosed to have neuroblastoma. We have come across 2 neuroblastoma families in which two out of the three siblings were affected in one family and two first cousins were affected in the second family. Cases have been reported in which offsprings have neuroblastomas and parents had ganglioneuroma(4,5). The pre-disposition to neuroblastoma in families has been reported to follow an autosomal dominant pattern of inheritance(6). According to Draper et al.(3) the occurrence of cancer in multiple members of a family can sometimes be accounted for by the existence of known genetic conditions predisposing to cancer. In other families however, the pattern of familial cancer might be attributable either to currently un-recognized cancer family syndromes or to exposure of family members to a common environmental hazard, or simply due to chance. In these 2 reported families, neither the patients nor their unaffected family members have had any exposure to a common environmental hazard. Moreover, no constitutional genetic syndrome or congenital anomaly that is consistently associated with predisposition to neuroblastoma were identified in those 2 families. Most of the diagnosis of familial neuroblastoma is made in the first 18 months of life, the median age of diagnosis is 9 months(7). This is in contrast with a median age of 22 months for neuroblastoma in general popula-tion. At least 20% of patients with familial neuroblastoma have bilateral adrenal or multi-focal primaries. In the present reported families, Patient I was diagnosed at the age of 9 months, but the other 3 patients ages approximate the median age at diagnosis in unselected patient, and are well beyond the peak age of diagnosis of familial and hereditary case (3 yr, 5 yr and 2� yr). Patient II had bilateral adrenal primaries which is more common in familial neuro-blastomas. With the important exception of multiple primaries clinical findings do not identify hereditary cases. Knudson and Strong(8) postulated a two stage genetic model for NB similar to that for retinoblastoma. In familial or hereditary cases of NB, the first hit is an inherited germline mutation present in all cells of body and 2nd hit occurs post zygotically in only the somatic target cell, the neuroblast. In sporadic non-hereditary cases, both mutations are postzygotic events in the same neuroblast. This theory confirm with the finding that the familial neural crest tumors tend to cluster in the earlier years of life. Knudson and Strong(8) estimated that as many as 22% of all neuroblastoma may be the result of a germ line mutation. Reports from the Childhood Cancer Research Group (CCRG)(4) showed that the risk for a sibling of a neuroblastoma patient of developing the same cancer is of the order of 1 per 1000. This figure implies that only a very small proportion of cases are transmitted from a parent, i.e., either the proportion of parents with germ cell mutation is low or that manifestation rate of potential genetic cases is low. Thus, for both Wilms tumor and neuroblastoma, the hereditary element has been suggested to be much smaller than originally suggested by Knudson and Strong(8). For families in which no clear genetic pattern emerges, some may be caused by an unknown genetic predisposition and some by shared environmental exposure, some are undoubtedly due to chance. Kushner and Helson(9) studied monozygotic twins con-cordant and discordant for neuroblastoma and this adds to the hypothesis that hereditary factors may predominate in neuroblastoma diagnosed in infants whereas nonheritable random mutational genetic events may be more important in neuroblastoma diagnosed after infancy. From data available so far, no specific gene locus is yet definite for familial neuroblastoma. This necessitates the need for examination of other candidate loci or even a genome wide search to identify the familial neuroblastoma predisposing gene. Genetic heterogeneity with several predisposing genes is yet another possibility that also has to be taken into consideration. References 1. Chatten J, Voorhees ML. Familial neuro-blastoma. N Eng J Med 1967; 277: 1230-1236. 2. Kushner BH, Gilbert F, Helson L. Familial neuroblastoma: Case reports, literature review and etiologic considerations. Cancer 1986; 57: 1887-1893. 3. Draper GJ, Sanders BM, Lennox EL, Brownhill PA. Patterns of childhood cancer among siblings. Brit J Cancer 1996; 74: 152-158. 4. Robertson CM, Tyrrel JC, Pritchard J. Familial neural crest tumors. Eur J Pediatr 1991; 150: 789-792. 5. Gerson JM, Chatten J, Eisman S. Familial neuroblastoma _ A follow up. N Eng J Med 1974; 290: 1487. 6. Knudson AG, Meadows AT. Developmental genetics of neuroblastoma. J Natl Cancer Inst 1976; 57: 675-682. 7. Buckley JD, Buckley CM, Breslow NE, Draper GJ, Roberson PK, Mack TM. Concordance for childhood cancer in twins. Med Pediatr Oncol 1996; 26: 223-229. 8. Knudson AG, Strong LC. Mutation and cancer. Neuroblastoma and pheochromocytoma. Am J Hum Genet 1972; 24: 514-532. 9. Kushner BH, Helson L. Monozygotic siblings discordant for neuroblastoma: Etiologic implica-tions. J Pediatr 1985; 107: 405-409.
|