|
|
|
Indian Pediatr 2015;52:
155-156 |
 |
White Matter Changes in GM1 Gangliosidosis
|
|
Moni Tuteja, *Abdul Mueed Bidchol, *Katta Mohan
Girisha and Shubha Phadke
From the Departments of Medical Genetics, Sanjay
Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar
Pradesh; and *Kasturba Medical College, Manipal University, Manipal,
India
Correspondence to: Dr Shubha R Phadke, Professor and
Head, Department of Medical Genetics,
SGPGIMS, Lucknow 226 014.
Email:
[email protected]
Received: July 14, 2014;
Initial review: October 08, 2014;
Accepted: December 08, 2014.
|
|
Background: GM1 gangliosidosis is
a disorder due to GLB1 gene mutation. Case characteristics:
A 4-yr-old boy with neuroregression and optic atrophy with
periventricular hyperintensity on magnetic resonance imaging.
Outcome: Beta galactosidase enzyme activity was low which was
confirmed by GLB1 sequencing. Message: We highlight the
white matter changes in late infantile GM1 gangliosidosis.
Key words: Convulsions, Magnetic resonance
imaging, Neuro-metabolic disorders.
|
|
GM1 gangliosidosis is a rare genetic disorder
caused by mutations in GLB1 gene leading to the deficiency of
enzyme beta galactosidase.[1]. The clinical manifestations are varied
due to accumulation of ganglioside in the lysosomes. It can be divided
into three types depending on the age of onset. Type 1 is an infantile
form, which presents between birth and 6 months of life. Type 2 is the
late infantile form, the onset varies between 6 months and 3 years of
age, the clinical features of which include neurological deterioration,
and cerebellar and extrapyramidal symptoms. Organomegaly, cherry red
spot and skeletal changes are usually not observed in this form. Type 3
(chronic/adult form) presents between 3 and 30 years. It is a gray
matter disease and diagnosis is confirmed by enzyme assay or mutation
detection. We present a patient with late infantile form of GM1
gangliosidosis.
Case Report
A four-year-old boy was brought with the complaint of
loss of all developmental milestones. He gained normal developmental
milestones till the age of 1 year. After 1 year, he had gradual loss of
all acquired skills. He had high-grade fever following which he
developed hypertonicity of the entire body, and then later at around 1½
years of age, he had unsteadiness of gait and had frequent falls. He was
bed-ridden since 2 years of age. There was a history of seizures with
onset at 1 ½ year of age, and apparent vision loss from the same age.
There was no history of any consanguinity in the family. Antenatal
period of the mother was uneventful. He was born by normal delivery with
no history of any postnatal or neonatal complications. There was a
history of similarly affected elder female sibling who died at 7 years
of age.
On examination, weight height and head circumference
were below -2SD. He was indifferent to the surroundings. There was no
startle response, no fixation to light, no nystagmus and no
hepatosplenomegaly. There was spasticity in the upper and lower limbs
and the deep tendon reflexes were absent. Fundus examination showed
bilateral optic atrophy. Nerve conduction velocity was normal and MRI
(magnetic resonance imaging) brain showed subtle T2 periventricular
hyperintensity in bilateral parieto- occipital regions extending to
frontal region. The subcortical white matter was also involved at places
(Fig 1a). There was thinning of corpus callosum with no
significant changes in basal ganglia, no evidence of cortical atrophy.
Ventricles were normal sized. Posterior fossa showed prominent cistern
magna and hypoplasia of inferior part of vermis.
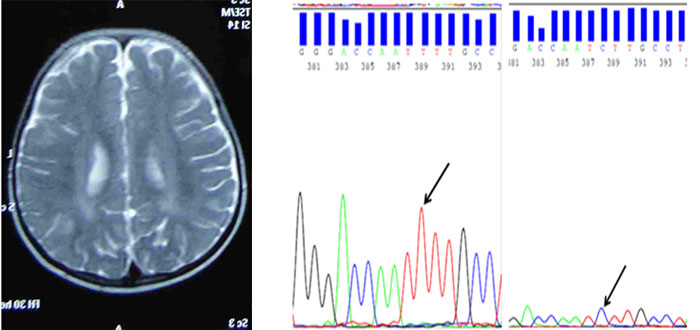
(a)
(b)
(c)
|
|
Fig. 1 (a) MRI brain showing mild T2
hyperintensities in the central periventricular as well as
subcortical white matter; (b) Sequencing result of wild type of
exon 9 of GLB1 gene; and (c) Sequencing result of patient
showing c.940T>C (homozygous) mutation in exon 9 of GLB1 gene.
(See color image at website)
|
With the above findings, Krabbe disease,
Metachromatic leucodystrophy and Neuronal ceroid lipofuscinosis were
considered as the most probable diagnosis. The enzyme activities for
palmitoyl protein thioesterase, tripeptidyl peptidase 1, beta
galacto-cerebrosidase and aryl sulfatase A were normal. There was
deficient beta galactosidase activity (2.1 nmol/hr/mg) (normal 70-324
nmol/hr/mg) which was tested as a control enzyme. The white matter
changes in MRI were not in the favor of GM1 gangliosidosis, which is a
gray matter disease. Hence, further GCMS (gas chromatography mass
spectrometry), TMS (tandem mass spectrometry) and lactate were also done
and were found to be normal. The enzyme assay for beta galactosidase was
repeated and showed deficient enzyme activity. The confirmation of the
diagnosis was done by sequencing of GLB1 gene (c.940T>C,
p.Phe314Leu (homozygous) mutation in exon 9 of GLB1 gene) (Fig.
1b and 1c). This was a novel mutation.
Bioinformatics analysis was conducted to access the
potential effect of this missense mutation on the protein; five
bioinformatics tools were used: the PolyPhen-2, SIFT, PROVEAN, Mutation
Taster and PANTHER. All the bioinformatics analyses predicted that
p.Phe314Leu is expected to be damaging to the protein function and hence
it is likely to be a disease-causing mutation. Parents were also
sequenced and they were found to be heterozygous for the same mutation.
Radiographs were not done as GM1 gangliosidosis was not suspected at the
initial evaluation.
Discussion
White matter abnormality in late infantile GM1
gangliosidosis have rarely been reported previously [2-4]. Moreover,
optic atrophy is a rare eye manifestation seen in this disorder [5-7].
Neuroimaging findings in late infantile GM1 gangliosidosis have been
rarely reported. Gururaj, et al. [2] reported MRI findings in two
infants with GM1 gangliosidosis and found delayed myelination and
abnormal appearance of the subcortical white matter, internal capsule,
and basal ganglia. Thalamic hyperdensity on CT scans and hypointense
signal of the thalami on T2-weighted MR images have also been reported
[3].
This case report highlights the MRI imaging and eye
findings in late infantile GM1 gangliosidosis which have been rarely
reported previously. This report broadens the phenotypic spectrum of
this disorder. Infantile GM1 presents with paucity of myelin in MRI and
the clinical and radiological picture of late infantile GM1 is entirely
different from infantile GM1 gangliosidosis.
Contributors: MT: clinical evaluation of child;
AMB: sequencing of GLB1 gene; KMG: analysis of sequencing; SP:
supervision and intellectual inputs. All authors contributed to
manuscript writing and its final approval.
Funding: ICMR, New Delhi; Competing interests:
None stated.
References
1. Sperb F, Vairo F, Burin M, Mayer FQ, Matte U, Giugliani
R. Genotypic and phenotypic characterization of Brazilian patients with
GM1 gangliosidosis. Gene. 2013;512:113-6.
2. Gururaj A, Sztriha L, Hertecant J, Johansen
JG, Georgiou T, Campos Y, et al. Magnetic resonance imaging
findings and novel mutations in GM1 gangliosidosis. J Child
Neurol.2005;20:57- 60.
3. Kobayashi, Takashima S. Thalamic hyperdensity on
CT in infantile GM1-gangliosidosis. Brain Dev. 1994;16:472-4.
4. Grandis E De, Rocco MD, Pessagno A, Venesseli E,
Rossi A. MR Imaging Findings in 2 Cases of Late Infantile GM1
Gangliosidosis. Am J Neuroradiol. 2009;30:1325-7.
5. Fei P, Qin M, Hu T. Ocular manifestations of
patients with gangliosidosis. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 1994;16:285-9.
6. Cabral A, Portela R, Tasso T, Eusbio F, Moreira A,
dos Santos HS, et al. A case of GM1 gangliosidosis type I.
Ophthalmic Paediatr Genet. 1989;10:63-7.
7. Sorcinelli R, Sitzia A, Loi M. Cherry-red spot,
optic atrophy and corneal cloudings in a patient suffering from GM1
gangliosidosis type I. Metab Pediatr Syst Ophthalmol. 1987;10:62-3.
|
|
|
 |
|
