|
|
|
Indian Pediatr 2015;52:
1029-1033 |
 |
Lysosomal Storage
Disorders in Indian Children with Neuroregression Attending a
Genetic Center
|
|
Jayesh Sheth, Mehul Mistri, Riddhi Bhavsar, Frenny
Sheth, #Mahesh Kamate,
*Heli Shah and
‡Chaitanya Datar
From the Department of Biochemical and Molecular
Genetics, FRIGE’s Institute of Human Genetics, FRIGE House, Satellite,
Ahmedabad; #Department of Pediatric Neurology and Child Development
Centre, KLES Prabhakar Kore Hospital, Belgaum, Karnataka; * Department
of Medicine, Sheth VS Hospital, Ellisbridge, Ahmedabad; and ‡Department
of Genetics, Sahyadri Medical Genetics and Tissue Engineering facility
(SMGTEF), Pune; India.
Correspondence to: Dr Jayesh J Sheth, Department of
Biochemical and Molecular Genetics, FRIGE’s Institute of Human Genetics,
FRIGE House, Satellite, Ahmedabad 380 015, India.
Email: [email protected]
Received: January 03, 2015;
Initial review: February 25, 2015;
Accepted: September 23, 2015.
|
Objective: To study the etiology of
neuroregression in children having deficiency of the lysosomal enzymes.
Design: Review of medical records.
Setting: Specialized Genetic Center.
Participants: 432 children aged 3 mo-18 y having
regression in a learned skill, selected from 1453 patients referred for
diagnostic workup of various Lysosomal storage disorders (LSDs).
Methods: Plasma chitotriosidase, quantitative and
qualitative glycosaminoglycans, and mucolipidosis-II/II screening
followed by confirmatory enzyme study using specific substrate was
carried out; Niemann-Pick disease Type-C was studied by fillipin stain
method on skin fibroblasts.
Results: Total 309 children (71.5%) were
diagnosed with different lysosomal storage disorders as the underlying
cause of neuroregression. Plasma chitotriosidase was raised in 82 of
135; 64 (78%) of these had various LSDs. 69 out of 90 cases showed high
excretion of glycoaminoglycans, and 67 (97.1%) of these were confirmed
to have enzyme deficiency for various mucoplysaccharide disorders. While
3/90 children with positive I-cell screening had confirmed mucolipidosis-II/III
disease. Among all, glycolipid storage disorders were the most common
(50.2%) followed by mucopolysaccharidosis (MPS) (21.7%) and sulphatide
degradation defect (17.5%). Neuronal ceroid lipofucinosis-1 & 2 (7.4%),
mucolipidosis-II/III (1%), Sialic acid storage disorder (1%), Niemann-Pick
disease type-C (1%) and Fucosidosis (0.3%) were observed with less
frequency. Most common phenotypes in all subjects were cherry red spot
(18.5%), hepatosplenomegaly (17.9%), coarse facies (15%), seizures
(13.1%) and skeletal abnormalities (12.14%).
Conclusions: Lysosomal storage disorders are
considered to be one of the common causes in children with regression in
learned skill, dysmorphic features and cherry red spot. Among these,
glycolipid storage disorders are the most common, followed by
mucopolysaccharidosis.
Keywords: Developmental delay, Glycolipid storage disorders,
Metabolic disorders, Mucopolysaccharidosis (MPS).
|
|
Neuro-regression in childhood could either be
genetic with neurometabolic origin or non-genetic causes such as
infections and toxins [1]. It has been observed that more than two-third
of the diagnosed cases of progressive neurological decline are due to
metabolic disorders [2]. Approximately 4.5% of the cases have
mitochondrial disease [3]
and several are found to have basic metabolic abnormalities like vitamin
B12 deficiency [4] and
thyroid disorders [5].
Lysosomal storage disorders (LSDs) are the heritable
group of nearly 40 heterogeneous disorders occurring due to genetic
defect in one or more specific lysosomal enzymes, activator protein or
membrane protein resulting in deficient enzyme activity [6-8]. There is
very little information available regarding the role of LSDs in
neuroregression, except for few studies demonstrating neurological
deterioration as the most commonly occurring pathophysiology of
lysosomal storage disorders in around one-third of the cases [9,10].
Though, individually these disorders are rare
(incidence 1:1,00,000), collectively they occur with the frequency of
approximately 1:7000-8000 live births [11-14]. Availability of prenatal
diagnostic facilities [15,16], newborn screening
[12,17] and the possibilities of early therapeutic
approaches [18,19] has increased awareness among medical fraternity for
different LSDs [20-23]. Therefore, we studied the frequency of various
LSDs as the cause of neuroregression in children from India.
Methods
This work presents the data on 432 children aged 3
months to 18 years referred to our institute between February 1997 and
May 2014, and selected from the cohort of 1453 patients referred for
various LSDs. Many of these children were also included in our previous
report on burden of LSDs in India [23]. They presented with regression
in learned skill with/without cherry-red spot, hepatomegaly/
hepatosplenomegaly, coarse facies, seizures, skeletal abnormality,
visual impairment and spasticity. Patients with neuroimaging findings
such as leukodystrophy, cerebral and/or cerebellar atrophy, white and
gray matter involvement were also included in the study. After obtaining
an Institutional ethics committee approval, an informed written consent
was obtained from the parents or the guardian while enrolling for the
previous study.
10-15 mL of random and/or morning void urine samples
were collected for screening, and confirmatory enzymes study was carried
on 6 mL peripheral blood collected in sodium heparin and/or EDTA
vaccutainers. A screening algorithm was used for plasma chitotriosidase
(ChT) [21] in 135 cases with hepatomegaly or hepatosplenomegaly and
neuro-regression. Urine glycosaminoglycans (GAG) screening [24] and
mucolipidosis-II/III (ML-II/III) [22] were carried out in cases with
coarse facial features and neuroregression (Fig. 1).
Confirmatory enzymes studies were carried out from leucocytes and/or
plasma using synthetic fluorogenic 4-methylumbelliferrone (4-MU) or
p-nitrocatechol sulphate (PNCS) substrates and enzyme activity was
expressed as nmol/hr/mg of protein, and for
b-Galactocerebrosidase
as nmol/17hr/mg protein [25]. The enzyme activity was carried out from
plasma in case of Sanfillippo type-B (MPS-IIIB) and ML-II/III, and was
expressed in term of nmol/hr/ml plasma [22,25].
For Niemann-Pick disease-C (NPD-C), skin
fibroblasts were cultured in lipid-deficient medium followed by fillipin
stain to confirm the presence of punctate granules [26,27].
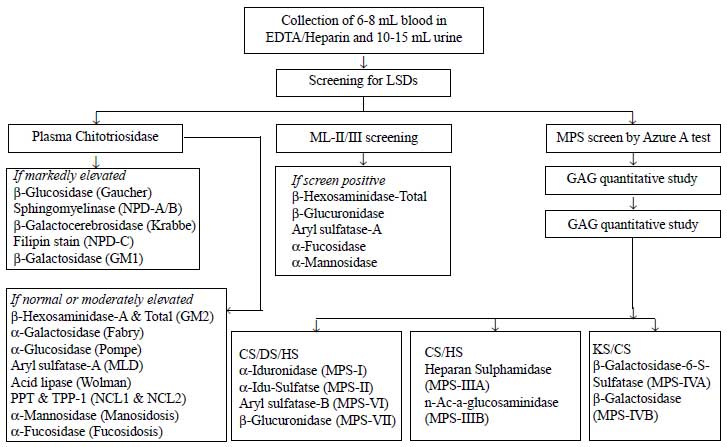 |
|
Fig. 1 Screening approach for various
lysosomal storage disorders.
|
Results
Out of 432 cases with the aforementioned clinical
signs and symptoms, 123 (28.5%) were found to be normal for lysosomal
enzyme activity and the rest were found to be affected with different
LSDs (Web Table I). Consanguinity among
parents was seen in 98 (22.7%) cases. The age of presentation for
diagnosis of storage disorder was 7 months to 7 years whereas late
juvenile presentation was seen in two cases of Niemann-Pick disease-B
(NPD-B), and Krabbe disease and Fucosidosis in one each. Adult onset
presentation was also seen in four cases of Sanfillipo type A/B
(MPS-IIIA/B) and two cases of Neuronal ceroid lipofucinosis type 2
(NCL2) and Metachromatic leucodystrophy (MLD).
Plasma chitotriosidase (ChT) screening of 135 cases
revealed raised ChT (106.9-30,000 nmol/hr/mL of plasma) in 82 (60.7%)
children. Enzyme study from leucocyte and/or plasma was carried out for
various LSDs and 78% (64/82) cases with raised ChT were found to have
Gaucher, Niemann Pick disease type A or B (NPD-A/B), Krabbe, GM1
gangliosidosis and Sandhoff disease, whereas 22% (12/53) with normal ChT
were affected with LSDs like Tay-Sachs, Neuronal ceroid lipofucinosis
type 1 and type 2 (NCL1 and NCL2), Metachromatic leukodystrophy (MLD),
NPD A/B and various mucopolysaccharide (MPS) disorders. Urine screening
for glycoaminoglycans (GAG quantitative and qualitative study) and
plasma screening for ML-II/III was carried out in 90 cases and 69 were
screen positive for urine GAG excretion suggesting presence of MPS
disorders with excretion of excess dermatan sulphate (DS) and moderate
heparan sulphate (HS) to mild chondroitin sulphate (CS) in 40 (57.9%)
and excess HS with mild CS in 27 (39.1%) cases. Further confirmation by
enzyme study was carried out in all patients. Moderate CS with mild HS
in 2 (2.9%) patients were found to be affected with ML-II/III and GM1
gangliosidosis one each. In I-cell screen, 87 cases were found to be
normal, while 3 patients were screen-positive; enzyme activity in plasma
further confirmed ML-II/III in all 3 screen- positive patients (Web
Table 1).
The most commonly diagnosed LSDs were in the group of
glycolipid storage disorders (50.16%) with GM2 gangliosidosis (23%), GM1
gangliosidosis (13.9%), Niemann Pick disease (12%) and Gaucher disease
(1.3%). (Web Table 1).
The common clinical phenotype observed among patients
with neuroregression affected with various LSDs were the presence of
cherry red spot (18.5%), hepatosplenomegaly (17.9%), and coarse facies
(15%) (Table I).
TABLE I Clinical Features of Children with Lysosomal Storage Disorders with Neuroregression (N=309)
|
Clinical Phenotype |
No. (%)(N=309) |
|
Cherry red spot |
58 (18.8) |
|
Hepatosplenomegaly |
56 (18.1) |
|
Coarse features |
47 (15.2) |
|
Seizures |
41 (13.3) |
|
Skeletal abnormality |
38 (12.3%) |
|
Cerebral and/or cerebellar atrophy |
23 (7.4) |
|
Psychosis |
20 (6.5) |
|
Leukodystrophy |
17 (5.5) |
|
Myoclonic jerks |
13 (4.2) |
|
Visual Impairment |
13 (4.2) |
|
Spasticity |
9 (2.9) |
|
White matter disease |
8 (2.6) |
|
Grey matter disease |
1 (0.3) |
Discussion
This is the largest data-set from India demonstrating
neuroregression in 29.9% of patients suspected with storage disorders,
71.5% of which had different types of LSDs, at a specialized genetics
center. The high occurrence of LSDs could be due to selection bias as
all referred cases were from the pediatric neurologists or pediatrician
or geneticist and it is highly likely that other causes of
neuroregression have been ruled out before suspecting for storage
disorders. Our study findings are in concordance with the Northern
Indian group demonstrating presence of LSDs in 69.2% of children having
neuroregression with highest frequency of mucopolysaccharidosis followed
by glycolipid degradation defects [10]. In a UK-based study where 40.4%
of children with progressive intellectual and neurological deterioration
(PIND) had LSDs with the highest frequency (31%) of NCL1 and NCL2 [9],
relatively large numbers of PIND cases were due to high rate of
consanguinity [9]. This is in accordance with our observation of 80% NCL
(1 and 2) cases from the region having 72% consanguinity.
The high proportion of glycolipid degradation defects
in this study, which is much higher than the previous study from India
[10], could be due to either the presence of founder mutation for
Tay-sachs disease in Gujarat [28] or referral bias due to lack of
investigative facility at other places in the region.
Mucopolysaccharidosis (MPS) was found to be the second most common LSD
with highest frequency of Sanfillipo disease (MPS-IIIA and IIIB). This
is in contrast to the reports of high number of MPS-I and -II in the
literature [10,23] and is likely to be due to overlapping phenotypic
features and limited diagnostic facility for these investigations in
most of the centers in the country. The third largest group of patients
with neuroregression were found with defects in sulphatide degradation
(17.5%) associated with MLD and Krabbe disease. This is almost similar
to what had been found by our group in an earlier study [23], while
Verma, et al. [10] have shown the presence of MLD in nearly 22%
of cases with neuroregression.
Major limitation of the present study is a referral
bias of children with neuroregression where previous workup for the
cause has been ruled out in the setting of lack of wider availability of
diagnostic facility at most of the places in the country.
To conclude, screening method for storage disorders
like mucolipidosis type II/III, MPS disorders and Gaucher/NPD-A/B have a
high predictive value for the confirmative diagnosis saving the
unnecessary cost of enzyme study. Additionally LSDs should be considered
to be one of the common causes of neuroregression in children with
regression in learned skill, dysmorphic features and cherry red spot.
Contributors: JS: study design, standardization
of technical procedure, preparation of manuscript and guarantor; MM:
processing the sample, analysis of data and preparation of manuscript;
RB: analysis of data and preparation of the manuscript; FS,HS,CD,MK:
critical evaluation of manuscript and patient management. All the
authors read and approved the manuscript.
Funding: Financial grant was provided by Indian
Council of Medical Research (ICMR) from the year 2006-2009 (Ref
54/2/2005-BMS) and 2010-1013 (Ref.54/1/2009-BMS).
Competing interests: Dr Jayesh Sheth is a
scientific adviser to Genzyme Sanofi India.
|
What is Already Known?
• Neuroregression is one of the common
observations in children with lysosomal storage disorders.
What This Study Adds?
• Screening by using biomarkers like Plasma
ChT, urine GAG and ML-II/III from plasma can provide the first
line diagnosis in children with suspected lysosomal storage
disorders.
|
References
1. Oscar-Berman M, Shagrin B, Evert DL, Epstein C.
Impairments of brain and behavior: the neurological effects of alcohol.
Alcohol Health Res World. 1997;21:65-75.
2. Tomas, Vila M, Vitoria MI, Gomez GF, Pantoja MJ,
Revert GM, et al. Epidemiology of progressive intellectual and
neurological deterioration in childhood- A multicentre study in the
Community of Valencia. Anales de Pediatría. 2013;78:303-07.
3. Verity CM, Winstone AM, Stellitano L, Krishnakumar
D, Will R, Mcfarland. The clinical presentation of mitochondrial
diseases in children with progressive intellectual and neurological
deterioration: a national, prospective, population-based study. Dev Med
Child Neurol. 2009;52:434-40.
4. Agrawal S, Nathani S. Neuroregression in vitamin
B12 deficiency. BMJ case reports 2009: bcr0620080235.
doi:10.1136/bcr.06.2008.0235.
5. Jain S, Chowdhury V, Juneja M, Kabra M, Pandey S,
Singh A, et al. Intellectual disability in Indian children:
Experience with a stratified approach for etiological diagnosis. Indian
Pediatr. 2013;50:1125-30.
6. Vellodi A. Lysosomal storage disorders. Br J
Haematol. 2005;128:413-31.
7. Futerman AH, van Meer G. The cell biology of
lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554-65.
8. Wilcox WR. Lysosomal storage disorders: the need
for better pediatric recognition and comprehensive care. J Pediatr.
2004;144:3-14.
9. Verity C, Winstone AM, Stellitano L, Will R,
Nicoll A. The epidemiology of progressive intellectual and neurological
deterioration in childhood. Arch Dis Child. 2010;95: 361-4.
10. Verma PK, Ranganath P, Dalal AB, Phadke SR.
Spectrum of Lysosomal storage disorders at a medical genetics center in
northern India. Indian Pediatr. 2012; 49:799-804.
11. Meikle PJ, Hopwood JJ, Clague AE, Carey WF.
Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-54.
12. Meikle PJ, Ranieri E, Simonsen H, Rozaklis T,
Ramsay SL, Whitfield PD, et al. Newborn screening for lysosomal
storage disorders: clinical evaluation of a two-tier strategy.
Pediatrics. 2004;114:909-16.
13. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE,
de Jong JG, Van WS, et al. The frequency of lysosomal storage
disease in the Netherlands. Human Genet. 1999;105:151-6.
14. Poupetova H, Ledvinova J, Berna L, Dvorakova L,
Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders
in the Czech Republic: comparison with data in different populations. J
Inherit Metab Dis. 2010;33: 387-96.
15. Lake BD, Young EP, Winchester BG.
Prenatal diagnosis of lysosomal storage diseases. Brain Pathol. 1998;8:
133-49.
16. Sheth J, Mistri M, Sheth F, Datar C, Godbole K,
Kamate M, et al. Prenatal diagnosis of lysosomal storage
disorders by enzymes study using chorionic villus and amniotic fluid. J
Fetal Med. 2014;1:17-24.
17. Mechtler TP, Metz TF, Muller HG, Ostermann K,
Ratschmann R, De Jesus VR, et al. Short-incubation mass
spectrometry assay for lysosomal storage disorders in newborn and
high-risk population screening. J Chromatogr B Analyt Technol Biomed
Life Sci. 2012;908:9-17.
18. Hoffmann B, Mayatepek E. Neurological
manifestations in lysosomal staorage disorders–from pathology to first
therapeutic options. Neuropediatrics. 2005;36:285-9.
19. Wang RY, Bodamer OA, Watson MS, Wilcox WR.
Lysosomal storage diseases: Diagnostic confirmation and management of
presymptomatic individuals. Genet Med. 2011;13:457-84.
20. Sheth J, Patel P, Sheth F, Shah R. Lysosomal
storage disorders. Indian Pediatr. 2004;41:260-5.
21. Sheth J, Sheth F, Oza N, Gambhir P, Dave U, Shah
R. Plasma chitotriosidase activity in children with lysosomal storage
disorders. Indian J Pediatr. 2010;77:203-5.
22. Sheth J, Mistri M, Kamate M, Vaja S, Sheth FJ.
Diagnostic strategy for Mucolipidosis II/III. Indian Pediatr.
2012;49:975-7.
23. Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A,
Godbole K, et al. Burden of lysosomal storage disorders in India:
experience of 387 affected children from a single diagnostic facility.
JIMD Rep. 2014;12:51-63.
24. Dembure, Philip P, Roesel AR. Screening for
mucopolysaccharidoses by analysis of urinary glycosaminoglycans.
Techniques in Diagnostic Human Biochemical Genetics: A Laboratory
Manual. Wiley-Liss, New York. 1991. p.77-86.
25. Shapria E, Blitzer MG, Miller JB, Africk DK. Fluorometric
assays in biochemical genetics: a laboratory Manual. New York, NY:
Oxford University Press.1989. p. 19-46.
26. Kruth, Howard S, Vaughan M. Quantification of low
density lipoprotein binding and cholesterol accumulation by single human
fibroblasts using fluorescence microscopy. J Lipid Res. 1980;21:123-30.
27. Sheth J, Sheth F, Oza N. Niemann-pick type C
disease. Indian Pediatr. 2008;45:505-7.
28. Mistri M, Tamhankar PM, Sheth F, Sanghavi D,
Kondurkar P, Patil S, et al. Identification of novel mutations in
HEXA gene in children affected with Tay Sachs disease from India.
PLoS One. 2012;7:e39122.
|
|
|
 |
|
