|
|
Case Reports Indian Pediatrics 2005; 42:1236-1239 |
||||||||||||||||||||||||||||
|
Detection of 22q11.2 Hemizygous Deletion by Interphase FISH in a Patient with Features of CATCH22 Syndrome |
||||||||||||||||||||||||||||
|
From Department of Reproductive Biology, Pediatrics* and Cardiology+, All India Institute of Medical Sciences, New Delhi 110 029, India. Correspondence to: Dr. Ashutosh Halder, Assistant
Professor, Department of Reproductive Biology, All India Institute of
Medical Sciences, New Delhi 110 029, India. Manuscript received: May 24, 2005; Initial review
completed: June 24, 2005;
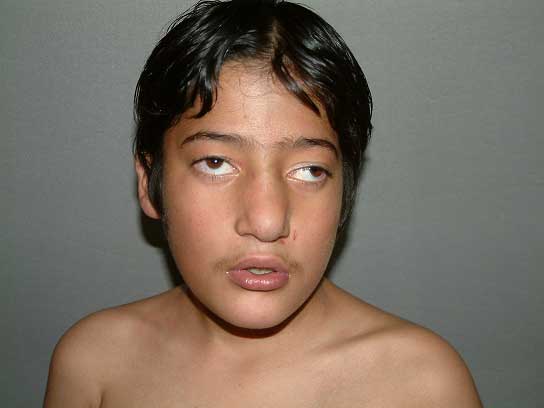
The 22q11.2 deletion syndrome is the most common microdeletion syndrome with an estimated incidence of one in 4000 births(1). Wilson, et al. (1993)(2) acronymed the common defects associated with this deletion as CATCH 22 syndrome (Cardiac abnormality, Abnormal facies, T cell deficit due to Thymic hypoplasia, Cleft palate and Hypocalcemia). Almost all cases of CATCH 22 syndrome result from 22q11.2 deletion. Diagnosis is based on prometaphase banding cytogenetics, FISH, array CGH, quantitative PCR and quantitative southern bloting(3-5). Chromosome 22q11.2 deletions are difficult to visualize even in case of high resolution banding, with a detection rate of ~20%(6). FISH is prime method for diagnosis. Demonstration of single signal in more than 10 metaphase spreads is the criteria for diagnosis(1). However, mitotic index with this syndrome is often poor(7) hence there is a need for interphase FISH. Diagnosis through interphase FISH on leukocytes(8) has been reported. Here we report rapid confirmation of diagnosis by interphase FISH using peripheral blood leukocytes first time from India. Case Report An 8-year-female child was referred from Doda, J & K, India to AIIMS for surgical correction of Tetralogy of Fallot. Cardiologist referred the case for genetic evaluation due to her dysmorphic features. She was home born at term to a 35 year-old para 5. There was no history of antenatal complications. Weight, length and head circumference at birth was not recorded. The baby had recurrent episodes of upper respiratory infection in infancy. Her milestone was delayed; started speaking at age of 3 and walking at age of five. She is going to nursery school since age of eight. She has dyspnea since last 5 years, which was gradual in onset but for last 2 years grade III with cyanosis. Her sibs were normal. There was no similar problem in the family. Physical examination revealed dys-morphic features, cyanosis, clubbing and sub-normal intelligence. She had broad bulbous nose, square shaped tip of nose, short filtrum, hypertelorism, telecanthus, squint and slanting eyes (Fig.1). She has low hair line. Her weight and height was normal however, she had relatively small head. Ears were low set and deficient in vertical diameter. Her hands and fingers were long and slender. She had nasal voice however there was no obvious cleft palate. She had moderate learning difficulties and intelligence quotient was 50% of normal for the age. Ophthalmologic examination was revealed squint (Fig.1) and hypoplastic optic disc in both eyes. Extensive cardiovascular work up including angiography and echo-cardiography was suggestive of Tetralogy of Fallot with reversal of flow (both valvular and infundibular pulmonary stenosis and right to left shunt). CT scan of head and brain was within normal. There was no hypocalcemia. Conventional cytogenetics was inconclusive due to poor quality and quantity of metaphase.
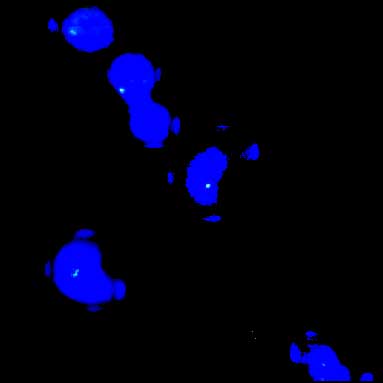
Since the patient had findings strongly suggestive of CATCH22 (diagnostic criteria; Table I) FISH study to detect a possible deletion was done. Interphase FISH was done using 1 ml blood from the patient and non-commercial probe obtained from Uniba Biologia, Bari, Italy. Interphase FISH result showed 98.5% nuclei with hemizygous deletion for 22q11.2 locus (Fig. 2). Table I
Discussion The proximal portion of human chromosome 22q has been implicated in the pathogenesis of CATCH22 syndrome; a clinically diverse group of conditions consisting DiGeorge Syndrome, Velocardio-facial Syndrome, Conotruncal Anomaly Face Syndrome, CHARGE Association as well as Isolated Conotruncal Heart Anomalies(9). In approximately 90% of patients the deletion size is 3 Mb whereas in remaining cases this varies from 1.5 to 2.5 Mb(10). No correlation is found between size of deletion and severity of symptoms. Chromosome 22 microdeletion occur de novo in most cases, however in 8% it may be inherited (50% risk for transmission). Early diagnosis is important because of its diverse medical complications and hereditary implications. Hence, there is a need for clinical diagnostic criteria in addition to a laboratory test for early, quick and comprehensive diagnosis of this syndrome. Retrospective examination of clinical data by Tobias, et al.(11) had revealed useful guidelines for clinical diagnosis of the condition (Table I). Authors suggested FISH analysis should be performed on patients who meet one of the criteria in column A. Any patient with a conotruncal cardiac anomaly, even in isolation, should be investigated for the presence of the deletion, because this problem occurs frequently (~50%). Alternatively, the possession or history of two features in column B or one feature in column B in addition to one in column C are regarded as sufficient to merit FISH investigation. These guidelines were devised with the aim of achieving high sensitivity in the initial detection of patients for whom the FISH analysis should be considered. Our case had one major (column A), three moderate (column B) and one minor (column C) criteria (Table I; marked bold) so warranted for confirmation by FISH. We performed FISH for detecting chromosome 22q11.2 microdeletion in peripheral blood uncultured leukocytes. This method has the advantage of a smaller sample requirement (100 µl blood is enough), quick (<24 hours) and free from tissue culture. As mosaicism rarely plays a role in this syndrome and as correct numbers of signals were seen in >98% of the cells, we think analysis of 100 cells is sufficient for diagnosis. In conclusion this result supports effectiveness of using clinical diagnostic criteria in combination with interphase FISH on peripheral blood leukocytes as a focused, two-steps method for rapid and reliable detection of 22q11.2 microdeletion. Acknowledgement Authors thank Professor Mariano Rocchi (University of Bari, Italy) for resources on molecular cytogenetics (clones for 22q11.2 locus). The authors also thank IRCH, AIIMS, New Delhi for providing fluorescent imaging facility. We are indebted to the clinician from Doda, J & K who referred the patient to AIIMS. Contributors: AH planned, organized and coordinated the work-up and drafted the paper; he will act as the guarantor of the paper. AF carried out FISH as per instruction and guidance of AH. He has done most of bench work. AH & MK had carried out clinical genetics work up. AH and AF was carried out conventional cytogenetic study. AS carried out cardiac workup and was in-charge of the patient. Funding: None. Competing interests: None. | ||||||||||||||||||||||||||||
|
References | ||||||||||||||||||||||||||||
|
![]()