|
|
|
Indian Pediatr 2016;53: 735-737 |
 |
Metatropic Dysplasia
with a Novel Mutation in TRPV4
|
|
Dhanya Lakshmi Narayanan,
#Gandham SriLakshmi Bhavani,
#Katta Mohan Girisha and Shubha R Phadke
From Department of Medical Genetics, Sanjay Gandhi
Postgraduate Institute of Medical Sciences, Lucknow; and #Department of
Medical Genetics, Kasturba Medical College, Manipal University, Manipal;
India.
Correspondence to: Dr Shubha Phadke, Professor and
Head, Department of Medical genetics, Sanjay Gandhi Post graduate
Institute, Lucknow 226 010, Uttar Pradesh, India.
[email protected]
Received: November 03, 2015;
Initial review: January 15, 2016;
Accepted: April 09, 2016.
|
Back ground: Metatropic dysplasia is a skeletal dysplasia
characterized by rhizomelia, severe kyphoscoliosis and a coccygeal tail.
Case characteristics: A 12 day-old male neonate had facial
dysmorphism, short limbs and coccygeal tail and showed radiological
features of metatropic dysplasia. Observation: A novel
heterozygous variant was observed in TRPV4 gene. Message:
We report a novel mutation in an Indian neonate with metatropic
dysplasia.
Keywords: Coccygeal tail, Genetics, Metatropic dysplasia,
TRPV4.
|
|
Metatropic dysplasia (OMIM 156530) is a severe
skeletal dysplasia characterized by rhizomelia, kyphoscoliosis and
coccygeal tail, manifesting in the immediate perinatal period [1]. Based
on the radiological findings and clinical features, three forms have
been described, including a lethal form, and a non-lethal form with less
severe radiographic manifestations. Recently, dominant mutations in
TRPV4 were identified both in severe and mild forms of metatropic
dysplasia by Camacho, et al. [2].
We are reporting a male neonate with features of
metatropic dysplasia, with a novel heterozygous mutation in TRPV4.
This is the first mutation-proven case from India.
Case report
A 12-day-old male new born was brought for evaluation
of dysmorphic facial features and short limbs. He was the first born
product of non-consanguineous marriage. Antenatal ultrasonogram was
normal. He was born at term, with uneventful perinatal period. He was
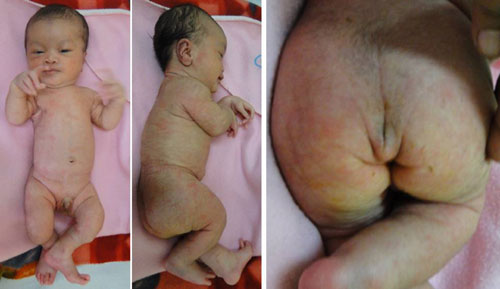
noticed to have short limbs compared to his trunk and a flat facies (Fig.
1). Parents were of normal stature.
His length was 43 cm (-3 SD), with an upper segment
to lower segment ratio of 1.8:1(Normal 1.7:1 at birth), indicating that
he had short limbs. His head circumference was 33.5 cm (normal).
Examination showed limitation of movements at both wrists, both knees
and both ankles, a prominent coccygeal tail and prominent knees, with a
gibbus in the thoracic spine. He had a flat facial profile (Fig.1).
 |
| (a) |
(b) |
(c) |
|
Fig. 1 Infant with dysmorphism showing
rhizomelia (a), flat facies (b), and coccygeal tail (c)
|
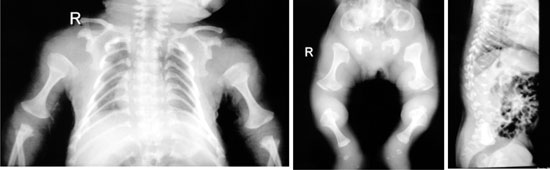
Radiographic evaluation showed severe platyspondyly
with wafer thin vertebra, dumb bell shaped long bones, halberd shaped
pelvis, narrow thorax and short ribs with cup shaped ends (Fig.
2).
 |
| (a) |
(b) |
(c) |
|
Fig. 2 X-rays of child showing dumb
bell shaped long bones (a,b), narrow thorax and short ribs with
cup shaped ends (a), Halberd shaped pelvis (b), platyspondyly
and wafer thin vertebral bodies (c).
|
Mutation analysis by sequencing all the coding
regions of TRPV4 gene (GenBank accession no. NM_021625) revealed
a novel heterozygous missense mutation, c.1834A>G (p.K612E), in exon 12
of TRPV4 gene. This missense variant was not present in dbSNP and
the 1000 Genome database and was predicted to be pathogenic by
pathogenicity predicting software Mutation Taster. The same mutation was
absent in parents.
Discussion
Our patient had short limbs, flat facies, prominent
joints, and a coccygeal tail, which helped us to establish the clinical
diagnosis of metatropic dysplasia. This condition was first described by
Maroteaux in 1966 [3]. The word ‘Metatropic’ refers to the change in
phenotype with short limbs and long trunk during neonatal period to
progressive short trunk during childhood, giving the name ‘changing
dysplasia’. There is a broad range of clinical severity in patients with
metatropic dysplasia [2]. The clinical features include short limbed
dwarfism, narrow cylindrical thorax and enlarged joints which present at
birth, with evolving severe kyphoscoliosis. The facial features are also
distinctive with squared-off jaw, prominent forehead and mid-face
hypoplasia [4]. Excessive ossification of the coccyx causing a prominent
"tail" has been described in patients [5]. Cervical myelopathy, odontoid
hypoplasia and spinal stenosis leading to neurological sequelae and
respiratory compromise causing death are the associated serious
complications [5]. Due to progressive kyphoscoliosis, pulmonary function
may become compromised.
Radiological findings include wafer-thin vertebral
bodies in newborn, halberd shaped proximal pelvis, brachydactyly with
delayed carpal ossification and flared proximal and distal metaphysis of
femora leading to ‘dumb bell shape’ of long bones [4]. Dumb bell-shaped
long bones are seen in other skeletal dysplasias like Kneist dysplasia,
fibrochondrogenesis and metatropic type of Spondylo epimetaphyseal
dysplasia (SEMD).
Till date, 33 mutation proven cases have been
described in literature [6]. Most of the mutations are missense
mutations. Based on the severity of the clinical features, the disease
is subclassified into a severe lethal form and a less severe non-lethal
form. There are eight allelic disorders, of which three are neurological
disorders, caused by mutations in TRPV4; These are Charcot Marie
Tooth Disease Type II C, scapuloperoneal spinal muscular atrophy and
congenital distal spinal muscular atrophy. This indicates that, TRPV4,
a cation channel which is selectively non-permeable to calcium,
encoded by the gene TRPV4, on chromosome 12, is involved in many
physiological processes [7]. It is now considered that TRPV4
mutations cause a phenotypic spectrum of skeletal dysplasias ranging
from mild brachyolmia to spondylometaphyseal dysplasia-Kozlowski to
metatropic dysplasia on the severe end of the spectrum [2]. No definite
genotype phenotype correlation could be observed [2], but degree of
activation of TRPV4 could determine the severity of the
phenotype.
Metatropic dysplasia is primarily a dominant
disorder. Even if the parents are unaffected, there could be recurrence
in siblings due to germline mosaicism in parents. To ascertain the
empiric risk, more experiments are needed [2]. But the risk of
recurrence is well below the 25% attributed for a recessive disorder.
Since the clinical and radiological features are
shared by other skeletal dysplasias, mutation analysis is essential to
reach a conclusive diagnosis. Prenatal diagnosis can be offered to
families where the mutation has been identified, by chorion villus
sampling and targeted mutation analysis of the fetus.
Acknowledgement: Indian Council of Medical
Research for funding (63/8/2010-BMS).
References
1. Beck M, Roubicek M, Rogers JG, Naumoff P, Spranger
J. Heterogeneity of metatropic dysplasia. Eur J Pediatr. 1983;140:231-7.
2. Camacho N, Krakow D, Johnykutty S, Katzman PJ,
Pepkowitz S, Vriens J, et al. Dominant TRPV4 mutations in
nonlethal and lethal metatropic dysplasia. Am J Med Genet A.
2010;152A:1169-77.
3. Maroteaux P, Spranger J, Wiedemann HR. [Metatrophic
dwarfism]. Arch Kinderheilkd. 1966;173:211-26.
4. Kannu P, Aftimos S, Mayne V, Donnan L, Savarirayan
R. Metatropic dysplasia: clinical and radiographic findings in 11
patients demonstrating long-term natural history. Am J Med Genet A.
2007;143A:2512-22.
5. Krakow D, Vriens J, Camacho N, Luong P, Deixler H,
Funari TL, et al. Mutations in the gene encoding the
calcium-permeable ion channel TRPV4 produce spondylometaphyseal
dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet.
2009;84:307-15.
6. Dai J, Kim OH, Cho TJ, Schmidt-Rimpler M, Tonoki
H, Takikawa K, et al. Novel and recurrent TRPV4 mutations and
their association with distinct phenotypes within the TRPV4 dysplasia
family. J Med Genet. 2010;47:704-9.
7. Vriens J, Watanabe H, Janssens A, Droogmans G,
Voets T, Nilius B. Cell swelling, heat, and chemical agonists use
distinct pathways for the activation of the cation channel TRPV4. Proc
Natl Acad Sci U S A. 2004;101:396-401.
|
|
|
 |
|
