|
|
Case Reports Indian Pediatrics 2005; 42:822-826 |
||||||
|
Acrodysostosis: Autosomal Dominant Transmission |
||||||
|
Ajai Perti* Grace Thomas From Departments of Pediatrics and *Radiology, Indira Gandhi Co-operative Hospital, Kadavanthra, Cochin 682 020, Kerala, India.
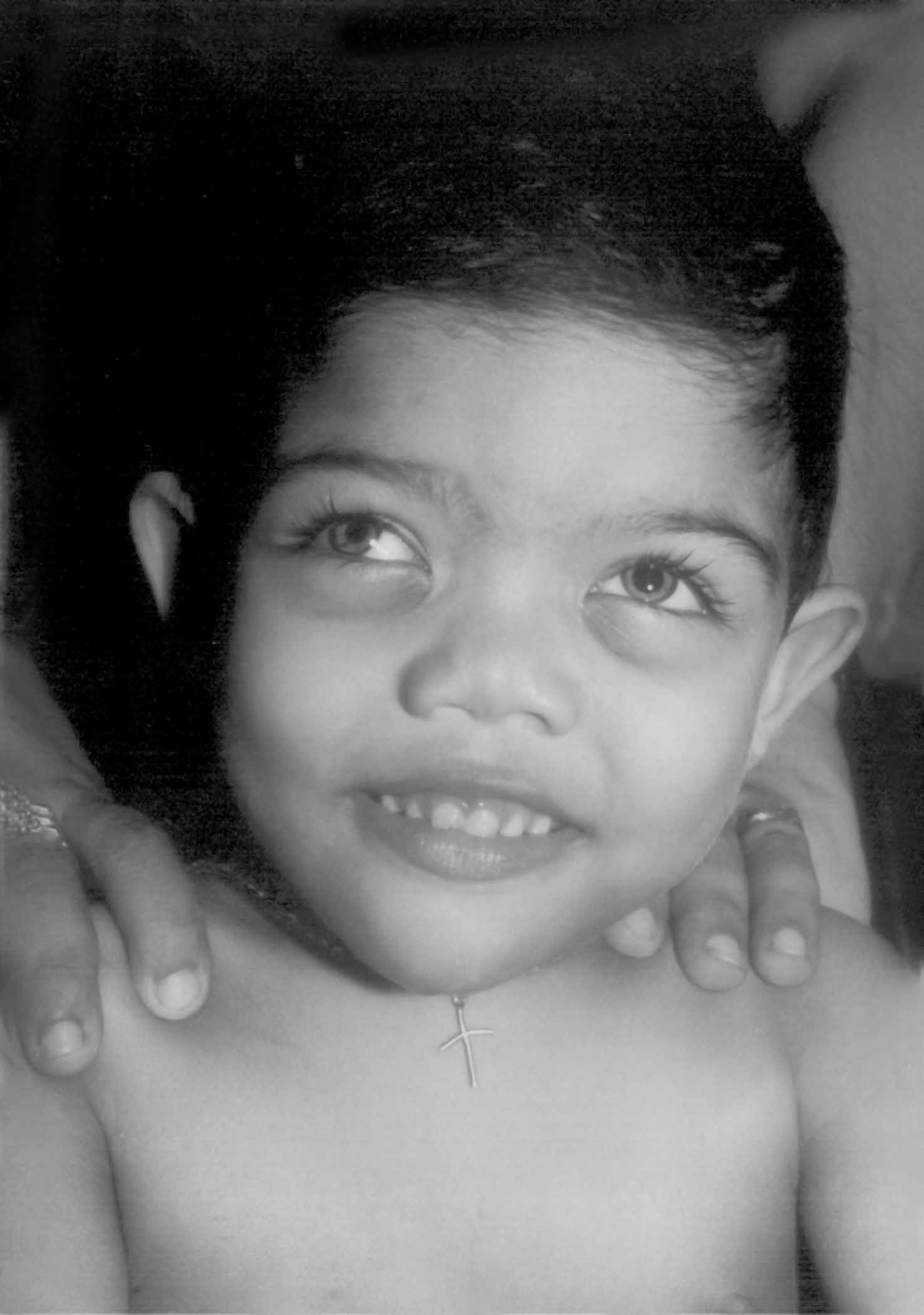
Acrodysostosis is a rare skeletal dysplasia, first described by Maroteaux and Malamut in 1968, and since then around 40 cases have been published in the literature(1). Though most case reports have occurred sporadically, it is believed to be an autosomal dominant condition. In this report we are presenting the first case with autosomal dominant inheritance pattern of acrodysostosis from India. Case Report A 2½-year-old male child with psycho-motor delay was referred for evaluation. He was born to a 24-year-old primigravida mother and a 37-year-old father and weighed 2500 g at birth. There was no history of consanguinity. A chromosomal evaluation showed a normal 46-XY pattern. He started to walk by 2 years of age and is presently able to run about and says 4-5 monosyllables. Evaluation by Washington’s scale showed a receptive language of 16 months and expressive language of 13 months. Height of the child was 88cm (<25th centile) and whereas weight (13kg )and head circumference (48.5 cm) were <50th centile. He had a dysmorphic facies with a broad and depressed nasal bridge and an upturned nose with anteverted nostrils. He had hypoplastic maxillae, a tendency to hold the mouth open, gingival hyperplasia, and dental malocclusion due to anterior open bite. Eyes showed bilateral heterochromia of iris and epicanthal folds and hypertelorism with a normal fundus (Fig. 1). The striking feature was the short stubby fingers with short and broad hands and wrinkling of the skin of the dorsum of the hands, and bilateral simian creases. The toes were also short but in contrast to that he had hypertrophied great toes bilaterally. The nails were broad on all fingers and toes.
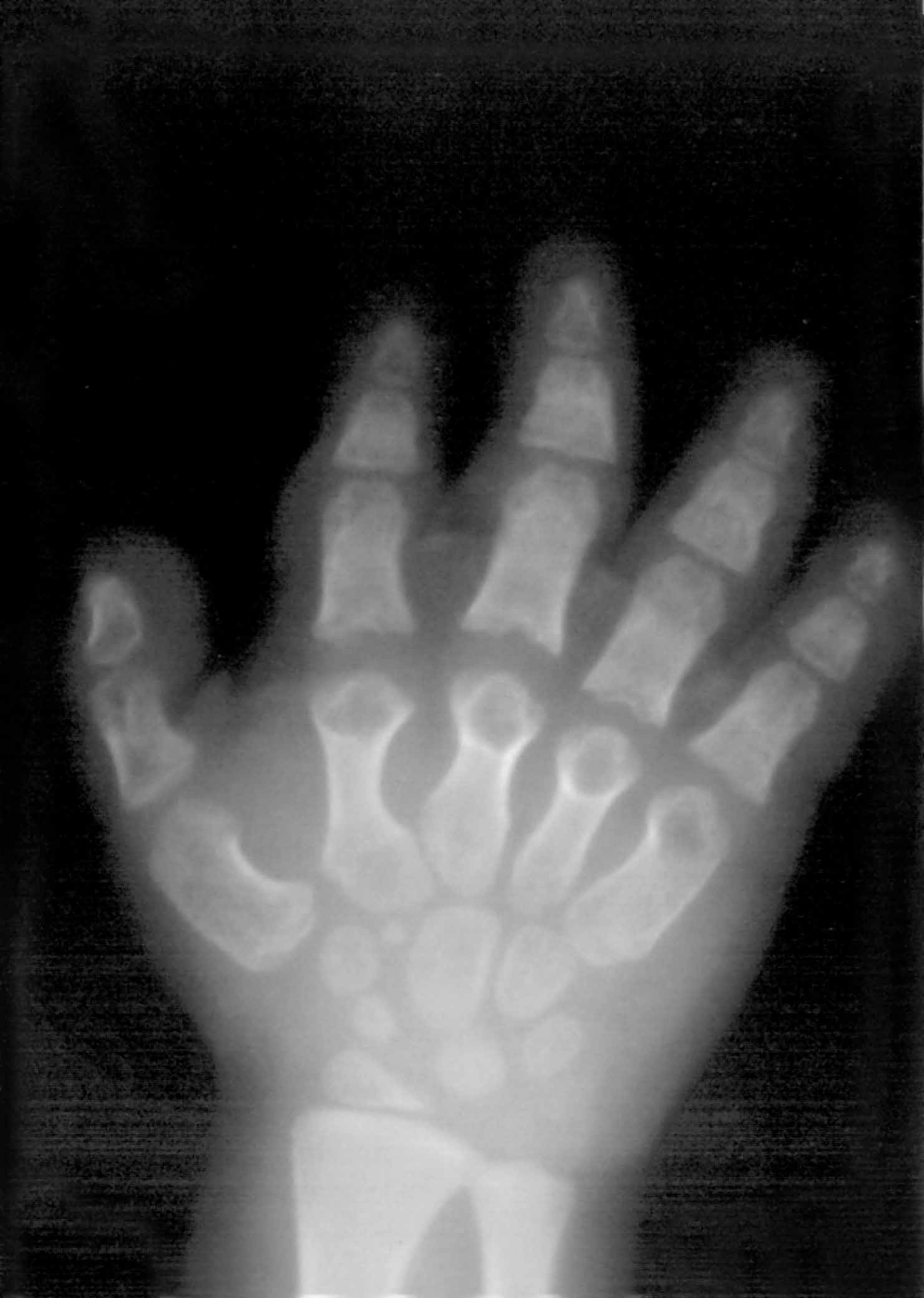
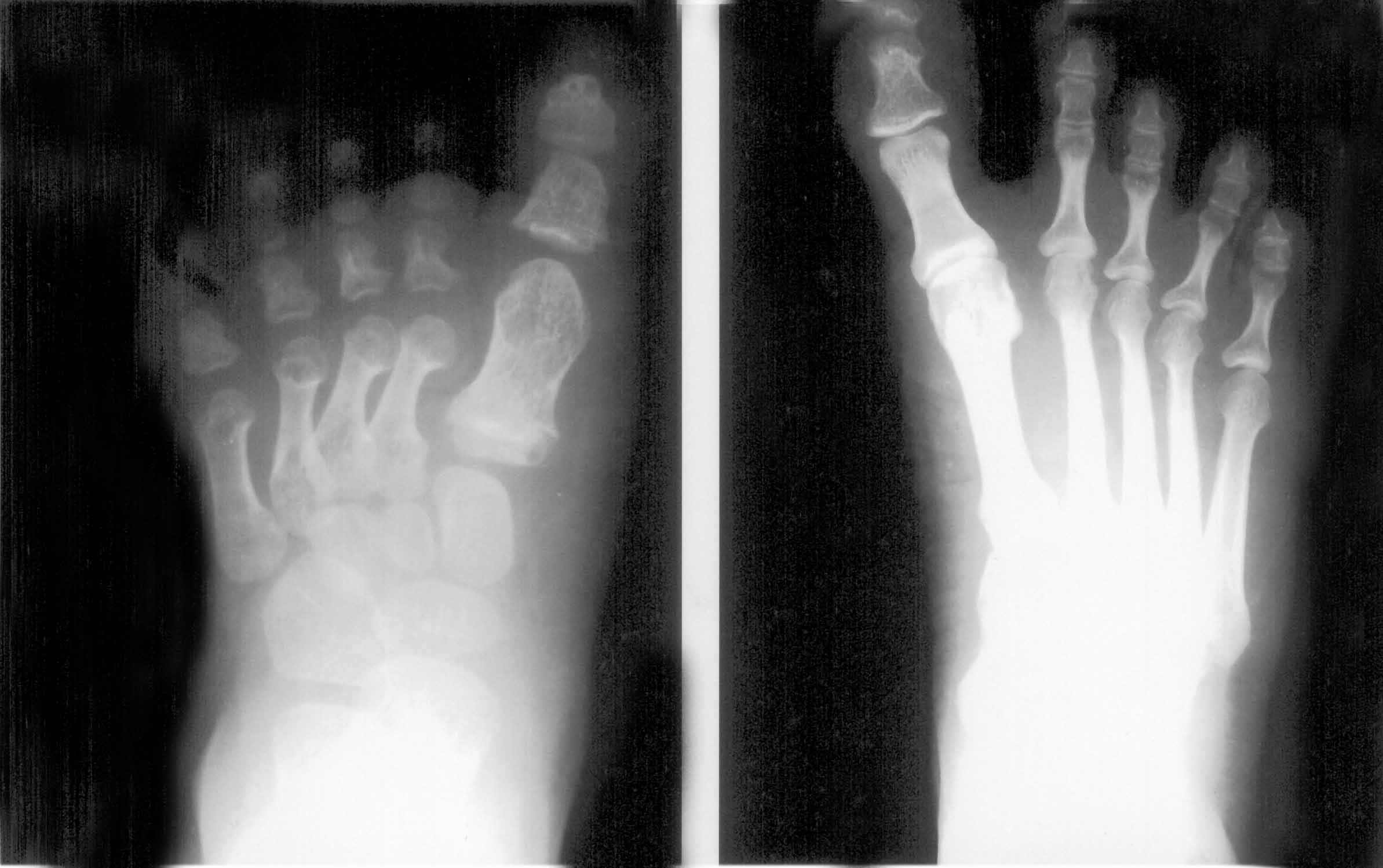
A detailed skeletal survey showed broad and short metacarpals and phalanges with cone-shaped epiphyses (Fig. 2). The bone age was significantly advanced, with 7 carpal bones on each side, with a bone age of more than 6 years and he had very large epiphyses of the long bones for his age. He also had significantly hypertrophied first metatarsals and phalanges of the great toes bilaterally confirming a first ray hyperplasia. The metatarsals and phalanges of the other toes were short and stubby. With these clinical and radiological features a diagnosis of acrodysostosis was made. A skull X-ray showed a very thick calvarium and increased mandibular angle 147.5º (129º mean value for <6 years) which are some of the less common features of this syndrome. Brain stem auditory evoked response was normal. Ultrasound study of abdomen showed normal kidneys, and MRI study of the brain and spine was within normal limits with no evidence of lumbar canal stenosis. Serum calcium, phosphorous and alkaline phosphatase levels were normal. Interestingly, the mother also showed a similar hypertrophy of the first metatarsals and phalanges of both great toes of her feet, thereby showing the autosomal dominant inheritance pattern with variable expressivity, as she did not have any of the other features of this syndrome (Fig. 3). No other family members had any similar anomalies.
The mother became pregnant a second time, and due to her fear of having a second child with psychomotor delay the fetus was closely monitored by serial 4 dimensional Echo evaluation with comparison of fetuses with similar gestational age and sex as controls for parameters like short fingers, hypoplastic nose and hyperplasia of the first metatarsal. Ultrasound study has its own limitations as nomograms are not available for detection of short fingers and nasal bone hypoplasia in acrodysostosis in fetuses. Fortunately the fetus did not show any of these features and the mother gave birth to a normal male baby. Discussion Acrodysostosis is a rare syndrome characterized by peripheral dysostosis (gross shortening of hands and feet), mental retardation, nasal hypoplasia (pug nose) and maxillary hypoplasia(2). Mental deficiency is reported in 77% of patients. The characteristic cranio-facial features include brachycephaly, low nasal bridge, anteverted nostrils, hypertelorism, epicanthal folds, tendency to keep the mouth open, prognathism, dental mal-occlusion, and delayed tooth eruption. The fingers and toes are abnormally short (peripheral dysostosis), with broad and short hands, with wrinkling of the skin of the dorsum of the hand. The nails on the fingers and toes are broad. Affected children exhibit advanced bone age and have large epiphyses for their age. Short stature and shortened hands and feet are caused by advanced bone age resulting in premature epiphyseal ossification. The metacarpals and phalanges show typical cone-shaped epiphyses, which fuse prematurely. Cone shaped epiphyses are a rare and distinctive clue to the diagnosis. Acrodysostosis, pseudohypoparathyroidism and acromesomelic dwarfism may present with cone shaped epiphysis. Even though these conditions share some important clinical features, advanced bone age and first ray hyperplasia are two important findings which distinguish acrodysostosis from the other conditions(3). A pointer in neonates with suspected acrodysostosis is the extensive epiphyseal stippling, which almost always disappears by 8 months of age(3). Vertebral defects include loss of normal caudal widening of the lumbar interpedicular distance, lumbar canal stenosis and scoliosis. Calvarial hyperostosis and increased mandi-bular angle are features that are less commonly associated with this condition. In our case the child had these two unusual features too. Urogenital anomalies include cryptorchidism, hypoplastic genitalia, irregular menses, hypogonadism and renal anomalies. Hearing deficit is described in 67% of patients but our patient had normal hearing. Most of these cases occur sporadically. Autosomal dominant inheritance pattern was seen in only 14% of patients in a review of 30 cases of acrodysostosis(4-6). In our case the mother had only first ray hyperplasia in both feet which is an important feature of this syndrome which suggests an AD inheritance with variable expressivity. Advanced paternal age has been noted in the fathers of some affected individuals(7). The differential diagnosis includes pseudohypoparathyroidism (Albright’s here-ditary osteodystrophy) which classically presents with short stature, short metacarpals and metatarsals and mental deficiency but they have low calcium and elevated phosphorus and alkaline phosphatase(8). Acromesomelic dysplasia also presents with extreme short stature, short hands and feet, and mild mental retardation, but our patient did not have mesomelia and was near normal in height. Lower thoracic kyphosis is an important clue to the condition, and is transmitted as an autosomal recessive pattern. Though patients with Robinow syndrome also have short hands and short stature and anteverted nostrils, their typical facies, hypoplastic genitalia and small penis differentiate them from acrodysostosis. Contributors: SSR diagnosed and managed the patient. AP did the radiological evaluation of the patient and was also involved in the drafting of the manuscript. GT was managing the patient during his infancy and had sent the patient for detailed evaluation. SSR will act as the guarantor of the manuscript. Funding: None. Competing interests: None.
| ||||||
|
References | ||||||
|
![]()