The diagnosis of a syndrome in a neonate with
multiple congenital malformations is very important from the point of
view of short and long-term management and genetic counseling. We report
a male neonate with Yunis-Varon syndrome, with some features not earlier
reported such as recurrence in successive offspring, polyhydramnios,
large testes and periosteal reaction and widening of ends of long bones.
Case Report
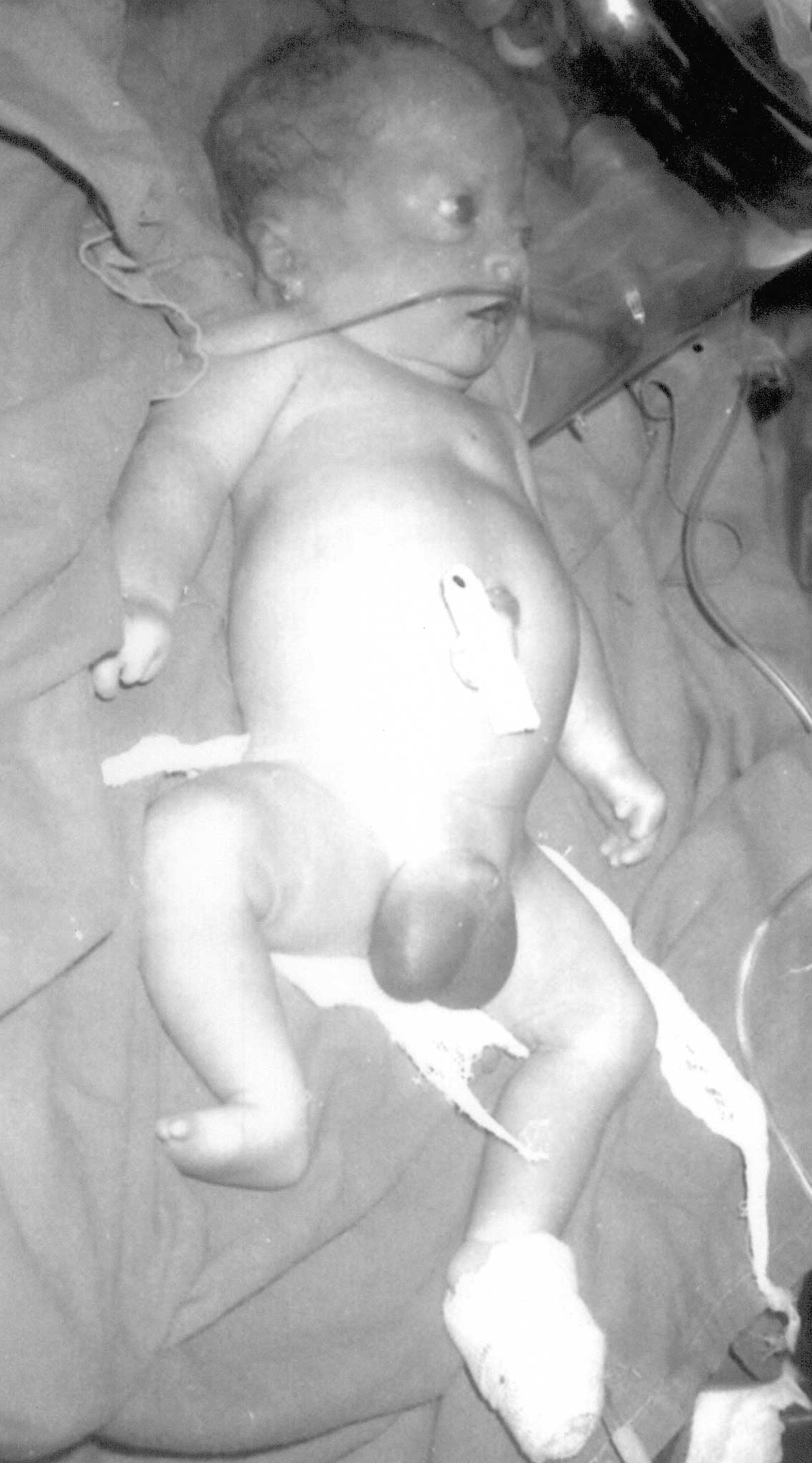
Male neonate weighing 2.6 kg was born at 36 weeks of
gestation of a non-consanguinous marriage by spontaneous vaginal
delivery to a 22-year-old gravida 2 mother. The mother gave history of
having given birth to an earlier baby with dysmorphic features who had
died on day 1 of life. Apart from polyhydramnios, the antenatal period
was unremarkable. The neonate required resuscitation at birth and
developed respiratory distress soon after.
Anthropometric data were in the 50th centile for
gestational age and upper: lower segment ratio was 1.9 : 1. The skull
bones were membranous, with wide, confluent fontanels and sutures. There
was sparse hair, absent eyebrows and eyelashes, high forehead,
anteverted nostrils, malformed ears, proptosis, hypertelorism, corneal
haziness and micrognathia. The thoracic cage was narrow and the nipples
were absent. Limbs were short with hypoplastic thumbs, absent terminal
phalanges and absent great toes. The scrotum was large and well formed
with a testicular volume of 6 mL bilaterally (Fig 1). The parents
confirmed that their previous offspring had similar malformations.
 |
|
Fig. 1 Absent nipples and toes, hypoplastic
fingers and thumbs and large scrotum. |
Radiological study revealed a poorly developed
calvarium, absent clavicles bil-aterally, small thorax, hypoplastic
thumbs and great toes and terminal aphalangia of both hands and feet.
Other findings included periosteal reaction of mid-shaft of the humeri
and femurs, narrow diaphysis and widened ends of tibiae and femurs. The
neonate was diagnosed as a case of Yunis-Varon syndrome.
The neonate was managed with ventilation and
supportive therapy, but expired at 10 hours of age. Autopsy ruled out
any additional malformations.
Discussion
Yunis-Varon syndrome is an extremely rare inherited
multisystem disorder with defects affecting the skeletal, ectodermal
tissue and cardio-respiratory systems. Less than sixteen cases have been
reported in the world literature. It is characterized by growth
retardation prior to and after birth; defective growth of the cranial
bones along with complete or partial absence of the clavicles (cleidocranial
dysplasia); characteristic facial features; and/or abnormalities of the
fingers and/or toes.
The first report of this condition appeared in 1980
when Yunis and Varon described 5 children from 3 families with
cleidocranial dysplasia associated with certain other dysmorphic
features including micrognathia, absent thumbs, distal phalanges of
fingers, and distal phalanx of the big toes, pelvic dys-plasia,
bilateral hip dislocation, and retracted and poorly delineated lips(1).
Two of the three sets of parents were consanguinous, suggesting an
autosomal recessive disorder. Subsequently, there have been isolated
case reports numbering a total of approximately fifteen cases with
absent or hypoplastic thumbs, short pointed fingers and toes and nail
hypoplasia or agenesis, being the invariable feature in addition to
cleidocranial dys- plasia, macrocrania and diastasis of cranial
sutures(3,4). Anteverted nostrils and short upper lip, microcephaly,
dislocated hips, hypoplastic great toes, syndactyly, dolico-cephaly,
micropenis, absent eyebrows and mental retardation have been frequently
described. Other associations reported have been tetrralogy of
Fallot(5), severe hearing impairment, pyloric stenosis(6), Dandy-Walker
malformation, hydrocephalus, and hypertension(7). The possibility of the
syndrome resulting from disordered lyso-somal storage was suggested by
Dworzak, et al. based on the finding of vacuolar myopathy on
muscle biopsy(8). Qualitatively abnormal bands for oligo saccharides and
neuraminic acid on urinanalysis by thin-layer chromatography and
prominent intraneuronal inclusions with vacuolar degeneration, mainly in
the thalamic nuclei, dentate nuclei, cerebellar cortex, and inferior
olivary nuclei, suggesting a defect of lysosomal storage were reported
by Walch, et al.(7).
Our patient, born of a non-consanguinous marriage was
the second offspring of the parents having similar physical features.
Although, our patient had no gross mal-formations of the organ systems,
poly-hydramnios, large testes and radiological features of widened ends
of tibiae and femurs and periosteal reaction in the long bones were
seen, which have not been reported earlier. Microscopic examination of
the internal organs did not show characteristic features suggesting a
storage disorder.
The prognosis of this syndrome is poor. Only three of
the 13 patients from the literature reviewed by Ades, et al.(5)
survived the first year of life, Two of the three survivors developed
severe physical and mental retardation(9), and one patient showed growth
retardation with normal intelligence.
Contributors: SB managed the case and drafted the
manuscript. RGH contributed to patient management and reviewed the
literature. He will act as guarantor.
Funding: None.
Competing interests: None declared.
